
INTRODUCTION
The rapid evolution of genomic technologies has transformed the landscape of animal breeding. High-throughput genotyping and sequencing provides breeders with access to vast amounts of genomic data and enables the precise selection of desirable traits [1]. These advancements have shifted traditional breeding methods to genomic selection, which leverages dense marker information to predict the genetic variants of individuals [2]. However, the success of genomic selection depends heavily on the accuracy and quality of the genomic data. Inaccurate or low-quality data can lead to inaccurate predictions that can compromise breeding programs and reduce their genetic gains [3]. Therefore, to ensure reliable predictions and maximize the potential of genomic selection, it is essential to implement stringent quality control (QC) measures at every stage of data processing.
Genomic data QC has several key components including the management of single nucleotide polymorphism (SNP) quality, the assessment of call rates, and filtering based on minor allele frequency (MAF) and Hardy-Weinberg equilibrium (HWE) [4]. High-quality SNP data is indispensable because errors in genotyping can lead to biased estimates of breeding values, which decreases the effectiveness of selection strategies [5]. Moreover, cost-effective low-density genotyping platforms often suffer from incomplete marker data so it is necessary to use imputation to deduce the missing genotypes [6].
QC processes are crucial for genomic selection, genome-wide association studies (GWAS), and population genetics analyses. These processes help ensure that the genomic data is accurate, reliable, and free from biases introduced by genotyping errors, population stratification, or other confounding factors [7,8]. This paper reviews QC strategies for genomic data and their applications in animal breeding programs. By examining various QC tools and methods, this paper aims to show the critical role that data integrity plays in achieving successful outcomes in genomic selection, GWAS, and population analyses [4,5].
GENOTYPING METHODS
WGS is a comprehensive method for analyzing the entire genome. Due to the decreased cost of sequencing and the ability to produce large amounts of genomic data, WGS has become a powerful tool for genomic research. SNP calling from WGS genomic data involves a series of critical steps to ensure accurate identification of genetic variants. The process starts with raw data preprocessing, where tools like FastQC evaluate the read quality [9]. This step is followed by trimming to remove adapters and low-quality bases by using either Trimmomatic or Cutadapt [10,11].
The cleaned reads are then aligned to a reference genome with BWA-MEM or Bowtie2 to generate SAM/BAM files [12,13]. These files are subsequently sorted, indexed, and processed to mark polymerase chain reaction (PCR) duplicates with Samtools, while the base quality scores are recalibrated using GATK [14,15]. Variant calling is performed using tools such as GATK’s HaplotypeCaller, FreeBayes, or Bcftools, which identify SNPs based on differences between the sequenced reads and the reference genome [15–17].
In post-calling, variants undergo filtering to remove false positives via GATK’s hard filtering or Variant Quality Score Recalibration (VQSR). The filtered SNPs are then annotated with functional information using tools like ANNOVAR or SnpEff [18,19]. Quality checks include the use of VCFtools for statistical analysis and IGV for visualization, and ensure the reliability of the called SNPs [16,20]. Joint genotyping across multiple samples and using population-specific reference panels are recommended to enhance the accuracy of SNP calling in WGS.
SNP arrays have significantly advanced genomic research in animal science by enabling the large-scale genotyping of SNPs. The development of SNP arrays began in the early 2000s to meet the demand for efficient and cost-effective methods to genotype large numbers of SNPs across the genome [21,22]. Early arrays marked a significant advancement by allowing simultaneous genotyping of thousands of SNPs, facilitating GWAS and the study of genetic variation in populations [21].
Over time, these arrays have evolved to include higher-density SNPs to improve coverage and accuracy, as seen in the Illumina BovineSNP50 array which has become a standard tool in cattle genomics [23,24]. Today, SNP arrays are essential for selecting desirable traits, estimating genetic merit, and managing inbreeding in animal breeding [1,2]. QC of SNP array data is crucial for ensuring accurate and reliable results, and involves assessing call rates, filtering based on MAF, and checking for HWE [4]. Tools such as PLINK and GenomeStudio are commonly used in these QC processes [5,25].
QC IN ANIMAL GENOMICS
MAF is a key metric in genetic studies. It represents the frequency at which the less common allele occurs in a given population. MAF is important for identifying rare variants which may not significantly contribute to overall genetic variation but can be crucial in specific contexts. MAF is calculated by determining the frequency of both alleles at a locus and taking the minimum of these two values. For example, if allele A has a frequency of 0.8 and allele a has a frequency of 0.2, the MAF would be 0.2. SNPs with very low MAFs, typically below 0.01 or 0.05, are often excluded from analyses because they may represent sequencing errors or lack statistical power in association studies [5].
Tools like PLINK and VCFtools [5,16] are widely used to calculate MAF, with PLINK’s --freq command being particularly popular [4]. In animal breeding, many researchers set threshold values for MAF to balance the need for sufficient variation while minimizing noise from rare variants. Typically, MAF thresholds in animal breeding studies range from 0.01 to 0.05 depending on the study’s objectives and the population structure being analyzed. For instance, a study on dairy cattle by Pryce et al. [26] and Kim et al. [27] sed a MAF threshold of 0.01 to ensure that the SNPs included were sufficiently informative for genomic predictions while also minimizing the influence of rare variants that might lead to spurious associations.
Call rate is another critical QC metric that measures the proportion of successfully genotyped samples for a specific SNP. A high call rate indicates that a SNP has been consistently detected across the sample population, while a low call rate may suggest issues with the genotyping process, such as poor quality or technical errors [7].
The call rate is calculated by dividing the number of successful genotype calls for a SNP by the total number of samples, then multiplying by 100 to express it as a percentage.
For instance, if 95 out of 100 samples have a successful genotype call for a SNP, the call rate would be 95% [4]. Normally, markers with a call rate less than 95% are removed, though other studies have set more stringent or lenient thresholds depending on the study design and objectives. For example, some studies have removed markers with a call rate below 99% to ensure extremely high data quality [28], while others have used a more relaxed threshold of 90% when working with larger datasets[29].
Tools like PLINK, SNP & Variation Suite (SVS), and GenomeStudio are widely used for calculating and filtering SNPs based on call rates because they offer robust functionalities for QC in genomic studies. PLINK is particularly popular due to its comprehensive command-line interface, where the --missing command calculates call rates at both the marker and sample levels, allowing researchers to easily filter out SNPs and samples that fall below the desired threshold [5]. SVS offers a user-friendly graphical interface and integrates various statistical tools, making it ideal for complex datasets and large-scale studies [30]. GenomeStudio by Illumina is another powerful tool specifically designed for managing and analyzing genotyping data with features for calculating call rates, identifying low-quality markers, and visualizing data for further inspection [25]. These tools are essential for ensuring that only high-quality data is used in subsequent analyses to improve the reliability of genomic outcomes.
HWE is a fundamental principle in population genetics. It states that allele and genotype frequencies in a population will remain constant from generation to generation in the absence of evolutionary influences [31]. Testing for HWE is an important QC step because deviations from this equilibrium can indicate issues such as genotyping errors, population stratification, or selection pressures [32]. To test for HWE, the observed genotype frequencies are compared to the expected frequencies under equilibrium conditions. For a biallelic SNP with alleles A and a, the expected genotype frequencies are p2 for AA, 2pq for Aa, and q2 for aa, where p and q represent the allele frequencies [33]. A chi-square test is commonly used to assess whether the differences between the observed and expected frequencies are statistically significant. Tools like PLINK and VCFtools are used to perform HWE tests [34]. SNPs that show significant deviation from HWE, typically with a p-value less than 0.001, are often excluded from analyses to prevent biases that could arise from genotyping errors or other confounding factors [4]. These QC metrics are foundational for ensuring high-quality genotypic data, forming the basis for accurate and reliable analyses in applications such as population analysis, GWAS, and genomic selection. Table 1 provides a summary of tools commonly used for QC steps, offering researchers practical options to streamline their workflows and enhance data integrity.
| Tools | Function | Reference |
|---|---|---|
| GEMMA | Application of linear mixed models and related models to GWAS | [4] |
| PLINK | Run association analyses and perform QC and regression steps | [5] |
| FastQC | Quality control checks on raw sequence data | [9] |
| Trimmomatic | Trim and crop FASTQ data | [10] |
| Cutadapt | finds and removes adapter sequences, primers, poly-A tails | [11] |
| BWA-MEM | produce multiple primary alignments for different part of a query sequence | [12] |
| Bowtie2 | aligning sequencing reads to long reference sequences | [13] |
| Samtools | Manipulate alignments in the SAM, BAM, and CRAM formats | [14] |
| GATK | Variant calling using sequencing data | [15] |
| VCFtools | Summarize, filter out, convert data into other file formats | [16] |
| FreeBayes | Bayesian genetic variant detector designed to fine SNPs | [17] |
| SnpEff | Annotation on genetic variants and predicts their effects on genes | [18] |
| ANNOVAR | Generate gene-based annotation | [19] |
| IGV | Visualization tool to simultaneously integrate and anlyze multiple types of genomic data | [20] |
| GenomeStudio | Normalize, cluster, and call genotypes | [25] |
| SVS | Perform analyses and visualizations on genomic and phenotypic data | [33] |
| BEAGLE | Genotype calling, phasing, and genotype imputation | [39] |
| Fimpute | Haplotype estimation or phasing and genotype imputation | [40] |
| Impute2 | Genotype imputation and haplotype phasing | [47] |
| Minimac | performs imputation with pre-phased haplotypes | [48] |
APPLICATION
Population analysis is invaluable for genomic studies in animal science because it enables researchers to assess the genetic structure, diversity, and evolutionary dynamics within and between populations. Accurately characterizing population structures is crucial for identifying subpopulations, measuring inbreeding levels, and understanding the genetic background of breeding populations, all of which are essential for maintaining genetic diversity and improving selection outcomes [35]. Tools such as PLINK, ADMIXTURE, and STRUCTURE are commonly employed to detect key characteristics for understanding the genetic landscape of animal populations, such as population stratification, admixture, and genetic differentiation [5,36]. For example, ADMIXTURE provides estimates of individual ancestry proportions. These estimates allow researchers to detect mixed genetic backgrounds that could influence trait analysis [36]. QC measures, such as filtering based on MAF, HWE, and genotyping call rates ensure the data used for population analysis is reliable [4,37]. MAF filtering helps exclude rare alleles that may introduce noise or result from genotyping errors [5]. Similarly, HWE filtering removes SNPs that deviate from expected frequencies due to selection or population substructures in order to prevent potential biases in the analysis [37]. Proper QC improves the accuracy of population structure analyses and mitigates the risk of confounding in subsequent analyses such as GWAS and genomic selection [4]. By accurately characterizing population structures, researchers can identify unique genetic markers and enhance their understanding of trait inheritance, and then design breeding strategies that optimize genetic gain and preserve diversity to support sustainable livestock production [35,36].
GWAS are powerful tools for identifying genetic variants associated with complex traits in animal breeding such as growth traits, disease resistance, reproductive traits, and carcass traits [2,4]. The reliability of GWAS findings hinges on rigorous QC procedures that ensure high-quality data throughout the process. This begins with careful study design and population selection, where potential confounders like population stratification are addressed through methods such as Principal Component Analysis (PCA) and linear mixed models to correct for genetic structure within the population [38]. Phenotype data must be accurately collected and screened for outliers to minimize noise. Genotype data undergoes thorough QC, including filtering SNPs based on call rates, MAF, and deviations from HWE [4,5]. For instance, SNPs with low call rates are excluded to avoid unreliable data that could lead to false-positive associations, while MAF filtering focuses the analysis on common variants that are more likely to have sufficient statistical power to detect true associations. HWE filtering is employed to remove SNPs that significantly deviate from expected allele frequencies because such deviations may indicate genotyping errors or underlying selection pressures [5]. To reduce redundancy and computational burden, linkage disequilibrium (LD) pruning is performed and missing genotypes are often imputed via reference panels using Fimpute or BEAGLE [39,40]. Tools like PLINK and GEMMA are widely used to implement QC measures and conduct association tests because they offer a robust framework for analyzing large genomic datasets [4]. Statistical analysis in GWAS is carried out using models appropriate for the trait under study, and corrections for multiple testing to mitigate the risk of false positives and meta-analysis may be employed when integrating results from multiple studies [41]. To ensure the robustness and high accuracy of the GWAS models, a 5-fold cross-validation is often used. In this method, the datasets are divided into five subsets. The model is iteratively trained on four subsets and tested on the remaining one to help validate the model’s accuracy and mitigate overfitting [42]. The results from GWAS offer valuable genetic variants for traits which can be targeted in marker-assisted selection and genomic selection programs. Genomic selection aims to ultimately improve the genetic merit of livestock populations [2]. The Fig. 1 summarizes the genotype QC workflow, with an emphasis on data preparation, QC steps, and their applications.
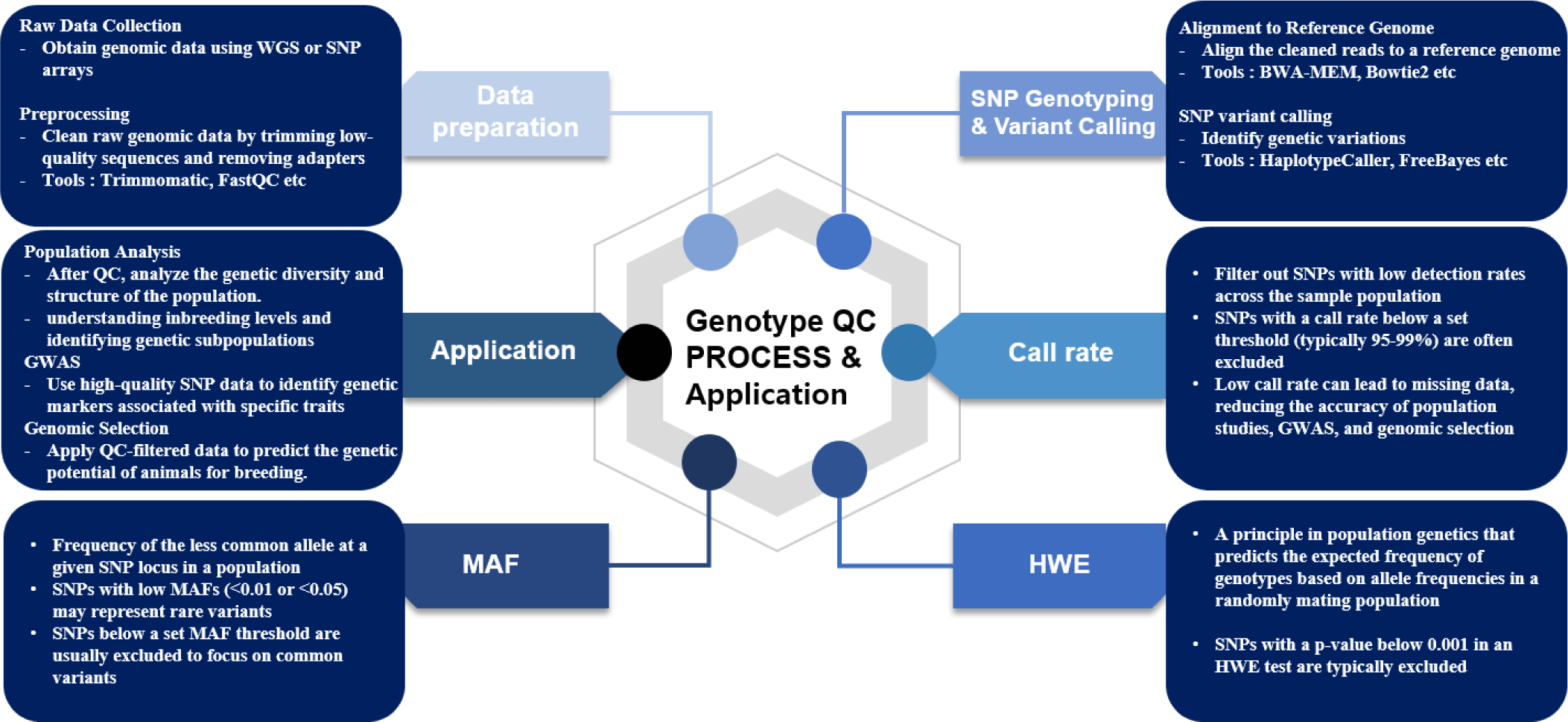
Genomic selection (GS) allows for the selection of animals based on SNP markers [43]. With the introduction of GS, animal breeding has dramatically advanced by overcoming the limitations of traditional selection methods like best linear unbiased prediction (BLUP) and marker-assisted selection [43,44]. GS relies on dense SNP data to estimate genomic breeding values, which are used to predict an individual’s genetic potential for economically important traits [2]. The accuracy of GS models is dependent upon the quality of the genomic data and the reliability of GS models can be enhance significantly by the inclusion of imputation methods to handle missing or low-density SNP data [45]. Imputation is beneficial in low-density platforms because it allows for the cost-effective use of genotyping while still leveraging the power of high-density SNP information. Imputation increases the accuracy of genomic predictions by inferring missing genotypes in order to improve the reliability of estimated breeding values even with fewer markers [6]. Several imputation tools, including FImpute [40], Beagle [39], Impute2 [46], and Minimac [47] are widely used in animal breeding to enhance the accuracy of GS models. Therefore, strict QC is essential [48]. QC methods, such as filtering SNPs based on call rates, MAF, and HWE, is critical to ensuring that the data is vigorous and reliable. High call rates are important because missing data can introduce bias and reduce the reliability of genomic estimated breeding values. Similarly, excluding SNPs with low MAF helps to avoid the noise associated with rare variants that may have little impact on prediction accuracy. Ensuring that SNPs conform to HWE expectations also prevents the inclusion of markers affected by selection, mutation, or other factors that could bias the GS models [4,5]. Advanced computational tools, such as genomic best linear unbiased prediction (GBLUP) and single-step BLUP (ssBLUP), and Bayesian methods (BayesA, BayesB, BayesC) integrate SNP effects across the genome to enhance the precision of breeding value predictions [49,50]. By using high-quality genomic data, GS enables breeders to make more accurate decisions that lead to faster genetic gains and the improvement of traits such as milk yield, growth rate, and carcass weight in livestock. This approach not only enhances the efficiency of breeding programs but also contributes to the long-term sustainability and productivity of animal populations [35].
CONCLUSION
High-throughput genotyping and sequencing has significantly advanced the field of animal breeding by enabling precise selection for desirable traits. However, the success of GS hinges on the accuracy and quality of the genomic data used. Rigorous QC measures are essential to ensure data integrity. These measures include SNP quality management, call rate assessment, and filtering based on MAF and HWE. These QC processes are crucial for GS, GWAS, and population genetics analyses. Implementing stringent QC strategies enhances the reliability of genomic predictions, which improves breeding programs and genetic gains. By maintaining high standards of data quality, researchers and breeders can make informed decisions that lead to sustainable and productive advancements in animal breeding.