













INTRODUCTION
Cultivated meat is meat produced through cell cultivation without the need to raise or slaughter livestock [1]. This offers potential advantages over livestock production, such as meeting the growing demand for meat protein sources as the world population increases [2,3]. Additionally, it has the potential to address issues related to climate change, ethics, religion, zoonotic diseases, and antibiotic resistance [4,5]. Traditional farm-based meat protein production will not be able to keep up with the rapidly growing world population. Cell culture-based meat production holds great potential as a replacement for meat protein [6,7]. As such, cultivated meat appears to be a promising solution for alleviating the growing demand for meat protein and addressing problems in the livestock industry. Meat primarily consists of skeletal muscle, which is composed of 90% muscle fibers and 10% connective and fatty tissues [8]. Cell types required to replicate meat with cultivated meat include skeletal muscle cells, adipocytes, and fibroblasts [9]. Among these various cell types, cultivated meat production mainly focuses on myosatellite cells (MSC) [10]. MSCs, also known as satellite cells, are located beneath the basement membrane of muscle fibers. They are typically in a quiescent state. However, they become activated by stimuli such as muscle damage [11,12]. MSC activated by muscle damage can proliferate in a myoblast state (with some returning to the quiescent state) and then develop into myotubes through intercellular fusion [13,14]. These MSC play an important role in muscle growth and regeneration [15]. Many research studies have been conducted on MSCs and methods for their isolation from livestock and maintenance in vitro have been well established [11,16–18]. To produce cultivated meat, activation of MSCs must be enhanced to increase cell proliferation and facilitate the creation of muscle tissue [19]. To achieve this, it is crucial to providing nutrients, hormones, and growth factors and to maintaining redox homeostasis in cells [20, –21].
Apis mellifera drone pupae is rich in chemical compositions, which contribute to high biological activities and nutritional properties of drone pupae [22]. Abundant proteins and vitamins in drone pupae can effectively prevent cellular aging [23]. In addition, drone pupae contain polyphenols and flavonoids with strong antioxidant properties, thereby reducing oxidative stress and contributing to disease prevention [24–26]. In addition to its antioxidant properties, drone pupae also exhibit various physiological activities, including antibacterial, antidiabetic, and anti-inflammatory effects [24,27,28]. Although drone pupae have a variety of functions, it is discarded on the grounds that it only consumes food without performing any functions other than mating with the queen bee [22,29,30]. It is easy to rear drone pupae since its production methods are well-established [31]. In addition, after being registered as a domestic food ingredient, drone pupae are being researched for the development of new promising functional ingredients with direct application to food [32]. In this regard, it is believed that utilizing drone pupae as a functional material could yield economic benefits.
Recent studies have suggested that applying insects or insect-derived substances to cells could play a role in regulating cell proliferation and differentiation [33–35]. As such, if drone pupae could be proven to have the ability to effectively promote proliferation and differentiation of Hanwoo myosatellite cells (HSC), they could serve as a functional material in cultivated meat production using HSC.
Studies on muscle growth using edible insects are still insufficient. Therefore, in this study, we hypothesized that drone pupae can be utilized as a functional material by improving the proliferation and differentiation of HSCs required for cultivated meat production and confirmed this.
MATERIALS AND METHODS
The amino acid analysis was requested from the Korea Food Research Institute. After placing approximately 0.2 to 5.0 g of drone pupae sample (Insect Seed Industry Research Institute of Chungcheongbuk-do Agricultural Research and Extension Services, Chungju) into a test tube, 10 mL of 6N HCl was added, and the mixture was stirred for 1 min. Oxygen was then removed from the test tube by filling it with nitrogen gas. The test tube was then immediately sealed with a stopper. The sample was hydrolyzed in a dry oven at 105°C for 22 h. After decomposition was complete, the sample was allowed to cool to 37°C. The sample in the test tube was then transferred to a 50 mL volumetric flask, added with deionized water to 50 mL, mixed, and filtered with a 0.2 μm PTFE membrane filter (Whatman , Maidstone, UK). From this, 1 mL was transferred to a 10 mL volumetric flask, added with 9 mL of deionized water, mixed, and filtered with a 0.2 μm PTFE membrane filter (Whatman) to prepare the test solution. This test solution was then analyzed with an L-8900 amino acid analyzer (Hitachi, Gunpo, Korea).
To analyze the content of vitamins B1, B6, B12, C, and E in drone pupae powder, a request for analysis was made to the department of Food Science and Technology at Chungbuk National University.
Vitamin B1 was analyzed using the method described by Jin et al. [36]. Approximately 2 g of the sample was treated with 50 mL of a 5 mM sodium 1-hexane sulfonate solution (composed of 7.5 mL acetic acid and 0.2 mL triethylamine per liter) and then subjected to ultrasonic extraction at 40°C for 30 mins. The extract was centrifuged at 20,000×g for 10 mins and the supernatant was then filtered through a 0.45 μm syringe filter (Whatman). Subsequently, it was subjected to high performance liquid chromatography-diode-array detection (HPLC-DAD). The mobile phases used for the analysis were 5 mM sodium 1-hexanesulfonate (solution A) and 100% methanol (solution B), with a flow rate of 0.8 mL/min. The column utilized was a YMC-Pack ODS-AM C18 (250 × 4.6 mm, 5 μm, YMC, Kyoto, Japan), and the analysis was conducted at a wavelength of 270 nm.
Vitamin B6 was analyzed using the method described by Lee et al. [37]. Five grams of the sample was added to 20 mL of 10 mM ammonium formate (0.1% formic acid). Ultrasonic extraction was then performed at 40°C for 30 mins. Afterward, the extract was centrifuged at 2,236×g for 15 mins at 0°C and the supernatant was collected. The above process was repeated once. The volume was then adjusted with a 10 mM ammonium formate (0.1% formic acid), which was used as the sample for HPLC analysis. This sample was subsequently analyzed using an HPLC (1200 series, Agilent, Santa Clara, CA, USA) equipped with a fluorescence detector (FLD, Agilent). The column used for the analysis was an Imtakt Scherzo SW-C18 (150 × 4.6 mm, 3 μm, Shiseido, Kyoto, Japan), with detection wavelengths set at Exλ = 290 nm and Emλ = 396 nm. The mobile phases consisted of 10 mM ammonium formate (0.1% formic acid, solution A) and 100% methanol (solution B), and the analysis was conducted at a flow rate of 0.7 mL/min.
Vitamin B12 was analyzed using the method described by Kwon et al. [38]. Five grams of the sample was treated with 0.5 mL of 1% sodium cyanide solution and 49.5 mL of sodium acetate buffer, followed by ultrasonic extraction for 10 mins. After extraction, the extract was placed in a 95°C water bath and shaken at 70 rpm for 1 h. The extract was then allowed to cool at room temperature, after which it was centrifuged and filtered. The filtrate was applied to an immunoaffinity column (EASI-EXTRACT® Vitamin B12, R-Biopharm, Glasgow, UK) to concentrate vitamin B12. The concentrated sample was then analyzed using an HPLC (5000 Chromaster, Hitachi, Tokyo, Japan) equipped with a UV detector (Hitachi), with detection at 361 nm. The column used for the analysis was an ACE 3 AQ (150 × 3.0 mm i.d., ACE, Scotland, UK), and the mobile phases consisted of ultra-pure distilled water (solution A) and 100% methanol (solution B).
Vitamin C was homogenized by adding 1-2 drops of 1-octanol to about 1 g of the sample. After adding 30 mL of extraction solvent, the mixture was homogenized using a homogenizer (AM-7, Nissei, Izumichom, Tokyo, Japan) for 2 min. The extraction solvent was prepared by dissolving 900 mL LC water, 50 g meta-phosphoric acid, 1.43 g TCEP, and 2 mL 500 mM EDTA. After its pH was adjusted to pH 1.55 with 8 N NaOH, it was filtered under reduced pressure. The homogenized sample was sonicated for 10 mins and then centrifuged at 1,853×g for 15 mins. The volume was adjusted to 50 mL with the extraction solvent. Subsequently, 1 mL of this solution was taken, filtered through a 0.45 μm syringe filter (Whatman), and analyzed using HPLC. The column used for the analysis was a Mightysil RP-18 column (4.6 × 250 mm, 5 μm). Detection was performed at 254 nm with a flow rate of 0.7 mL/min, and a column temperature of 40°C. The mobile phases were prepared as follows: A (0.05% formic acid in water) and B (0.05% formic acid in acetonitrile). For the quantitative analysis, a standard solution of L-ascorbic acid was prepared by dissolving 0.05 g in a 50 mL volumetric flask, and then diluted to make a 1,000 µg/mL standard solution, which was subsequently further diluted for use.
Vitamin E analysis was conducted using the method described by Kwon et al. [39]. Four grams of the sample was treated with 20 mL of ethanol containing 6% pyrogallol and filled with nitrogen gas. Then 8 mL of 60% potassium hydroxide (KOH) was added to induce a saponification reaction in a constant temperature water bath at 75°C for 50 min followed by cooling. To the reaction solution, 30 mL of 2% sodium chloride (NaCl) and 20 mL of hexane containing 0.01% butylated hydroxytoluene (BHT) were added. The extraction process was then repeated three times. Any residual moisture was removed. This extract was dissolved in a 50 mL flask as an extraction solvent. The solvent was removed under nitrogen, re-dissolved according to the concentration of the test solution, filtered through a 0.45 μm PTFE membrane filter (Whatman), and analyzed by HPLC. The analysis was performed using an HPLC system with a solvent delivery pump and an FP2020 fluorescence detector. The column used was a LiChrospher Diol 100 (5 μm, 250 × 4 mm, Merck, Kenilworth, NJ, USA). The excitation wavelength of the fluorescence detector was set to 290 nm and the emission wavelength to 330 nm. The mobile phase consisted of n-hexane containing 1.1% isopropanol, with a flow rate of 1.0 mL/min. Tocopherols (α-, β-, γ-, δ-) and tocotrienols (α-, β-, γ-, δ-) were used as standard substances. Each standard substance was identified by comparing its retention time, and a calibration curve was established by plotting peak areas against the concentrations of the standards to confirm linearity and correlation (R²) for quantification.
The mineral analysis was requested from the joint laboratory at Chungbuk National University. Contents of minerals in drone pupae powder, specifically calcium (Ca), copper (Cu), iron (Fe), potassium (K), magnesium (Mg), manganese (Mn), sodium (Na), zinc (Zn), and phosphorus (P), were quantitatively analyzed. For this purpose, we utilized an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, Spectro ARCOS, Kleve, Germany) at Shared Laboratory Facilities of Chungbuk National University (Chungju, Korea). To approximately 0.2 g of the sample, 6 mL of nitric acid was added. Approximately 0.2 g of the sample was taken for analysis and treated with 6 mL of nitric acid using a Microwave Digestion System (ultraWAVE, Milestone, Shelton, CT, USA), followed by dilution to a final volume of 50 mL. Processed samples were then diluted 10-fold and 100-fold before analysis with the ICP-OES. Calibration curves were established using standards at different concentrations (0, 0.02, 0.1, 1, 10, and 100 mg/kg). Calibration curve selection and recalculations were performed based on ICP-OES results to obtain final concentrations.
The process for preparing drone pupae extract powder (DEP) is shown in Fig. 1. Fifty grams of drone pupae powder were mixed with 500 mL of deionized water. The container was sealed with foil and stored in a refrigerator at 4°C for 72 h. The mixture was then filtered through a whatman No. 2 filter paper (Advantec®, Tokyo, Japan). The filtered extract was frozen at −50°C for over 24 h and subsequently lyophilized in a freeze-dryer (FDU-1110·2110, Sunil Eyela, Sungnam, Korea) for 72 h. The dried extract was pulverized using a mortar and pestle and stored at −80°C until used in experiments.

Hanwoo myosatellite cells were seeded in 96-well plates pre-coated with collagen at a density of 15,000 live cells per well and cultured at 37°C in an incubator with 5% CO2 until confluent was reached. Upon achieving confluent, cells were treated with various concentrations (0, 10, 100, 200, 400 µg/mL) of DEP for 24 h. Subsequently, cells were exposed to 700 µM H2O2 for 1 h. Cell viability was assessed by measuring absorbance at 490 nm using the MTS assay.
This analysis was conducted using the method described by Park et al. [40]. Rump muscle tissue was harvested from a 33-month-old Hanwoo steer at Farmstory Hannaeng (Eumseong, Korea). The collected tissue was transported to the laboratory in an ice box. Subsequently, HSC were isolated using collagenase type II at 600 units/mL DMEM (Cat # LS004176, Worthington Biochemical, Lakewood, NJ, USA) and harvested. Harvested HSC were stored in liquid nitrogen using a freezing medium (Cell Culture Freezing Medium, Cat # 12648-010, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and considered as passage 0. Some HSC were cultured and then analyzed by FACS with the following antibodies: APC anti-human CD29 antibody (1:10, Cat # 303008, BioLegend, San Diego, CA, USA), PE-Cy™7 anti-human CD56 (1:10, Cat # 335826, BD, Franklin Lakes, NJ, USA), FITC anti-sheep CD31 (1:10, Cat # MCA1097GA, Bio-Rad Laboratories, Hercules, CA, USA), and FITC anti-sheep CD45 (1:10, MCA2220GA, Bio-Rad). These HSC were sorted into CD31-/CD45-, CD29+/CD56+ populations using a FACS Aria II Cell Sorter (BD Life Sciences, San Jose, CA, USA). In this experiment, passage 2 HSC were utilized.
For proliferation culture, surfaces were coated with collagen. The collagen coating solution was made by diluting 1 M acetic acid (Cat # A0052, Samchun Chemicals, Seoul, Korea) in deionized water to achieve a final concentration of 2%, followed by diluting a 5 mg/mL bovine collagen type I solution (Cat # A1064401, Gibco) to a concentration of 0.5%. This solution was dispensed into a 96-well plate (Cell Culture Plate, SPL Life Science, Seoul, Korea) and left in a 37°C incubator for at least 16 h. Before experimental use, the coating solution was removed and wells were washed twice with 1X phosphate-buffered saline (PBS; Cytiva HyClone™, Logan, UT, USA) and air-dried. The medium (growth medium, GM) used was Ham’s F-10 medium (11550-043, Gibco) supplemented with 20% fetal bovine serum (FBS; 35-015-CV, Corning , NewYork, NY, USA) and 1% penicillin-streptomycin-amphotericin B (PSA; 17-745E, Lonza, Basel, Switzerland). DEP was added to the prepared GM at varying concentrations (0, 10, 100, 200, and 400 µg/mL) as shown in Table 1. To promote cell proliferation and growth, basic fibroblast growth factor (bFGF) was also added to achieve a final concentration of 0.05%.
| Trait | CON1) | D10 | D100 | D200 | D400 |
|---|---|---|---|---|---|
| DEP (μg/GM 1 mL) | - | 10 | 100 | 200 | 400 |
Cell viability was determined using trypan blue staining (T8154, Sigma, Gillingham, Dorset, UK). Cells were counted using an automated cell counter (Countess cell FL automated cell counter, Invitrogen, Waltham, MA, USA). For the proliferation culture, HSC were seeded at a density of 1,800 live cells/cm² into a 96-well plate (Cell Culture Plate, SPL Life Science) and cultured in an incubator at 37°C with 5% CO₂ for five days. The proliferation of HSC during the culture period was monitored using a microscope (EVOS-5000).
For differentiation culture, surfaces were coated with Matrigel. The coating solution was prepared by diluting Matrigel (Matrigel® Basement Membrane Matrix, Corning®, Herndon, VA, USA) with cold 1X PBS at a ratio of 1:200. This mixture was dispensed at 3 mL per T25 flask and 50 µL per well of a 96-well plate (black plate 33396, SPL Life Science). Plates were then incubated at 37°C for at least 4 h. Before use in experiments, the coating solution was removed, and the surfaces were washed once with 1X PBS and air-dried. The medium (differentiation medium, DM) used was prepared by supplementing DMEM (11995-065, Gibco) with 2% FBS and 1% PSA. DEP was added to the prepared DM at concentrations of 0 and 100 µg/mL, as indicated in Table 2.
| Trait | CON1) | D100 |
|---|---|---|
| DEP (μg/DM 1 mL) | - | 100 |
Cell viability was determined using trypan blue staining (T8154, Sigma) and cells were counted using an automated cell counter (Countess cell FL automated cell counter, Invitrogen, Waltham, MA, USA). Initially, cells in GM at 5,000 live cells/cm² were seeded into T25 flasks or at 10,000 live cells/cm² into 96-well plates (black plate 33396, SPL Life Science). Cells were cultured at 37°C with 5% CO₂ for 5 days in T25 flasks or 3 days in 96-well plates. Once cells became confluent, the GM was removed from both T25 flasks and 96-well plates and replaced with DM, followed by further incubation for 3 days in T25 flasks or 4 days in 96-well plates. During the culture period, the differentiation of HMC was monitored using a microscope (EVOS-5000).
Hanwoo myosatellite cells were cultured in 96-well plates (Cell Culture Plate, SPL Life Science). Their proliferation capacity was performed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). This utilized the reduction of the tetrazolium compound, MTS to formazan by dehydrogenase enzymes present in viable cells. After removing culture medium, MTS reagent was added to wells. Plates were incubated at 37°C with 5% CO2 for 2 h. Subsequently, absorbance was measured at 490 nm.
After 4 days of differentiation, the culture medium was removed from HSC cultured in 96-well plates (black plate 33396, SPL Life Science ) and cells were washed with 1X PBS. Cells were then fixed with 2% paraformaldehyde (PFA; in PBS) at 37°C for 45 mins, washed with 1X PBS twice, and permeabilized with 0.1% Triton-X (in PBS) at room temperature for 20 mins. Cells were then blocked with 2% bovine serum albumin (BSA) at room temperature for 30 mins, followed by two additional PBS washes. Mouse Monoclonal Anti-Myosin antibody (1:100, Sigma, Cat # M4276) was applied and cells were incubated at 4°C overnight. After overnight incubation, cells were washed twice with 0.05% Tween 20 in PBS (Cat # 1706531, Bio-Rad) and then incubated with a secondary antibody (Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488; 1:2,000, Catalog # A21121, Invitrogen) at room temperature for 30 mins. Finally, cells were washed with 0.05% Tween 20 and stained with Hoechst 33342 (1:2,000, Catalog # H3570, Invitrogen) at room temperature for 2 mins. Cells were washed twice more with 1X PBS and stored shielded from light.
Immunofluorescence-stained HSC were observed using a microscope (EVOS-5000). Images were obtained from nine zones per well. The procedure was repeated across five wells. All images were analyzed using ImageJ software (NIH, Bethesda, MD, USA) to measure fusion index (%) and myotube area (μm). Myotubes were classified as elongated structures containing more than two nuclei within a single membrane. The fusion index was calculated as the percentage of nuclei within myotubes relative to the total number of nuclei.
After HSC were differentiated for 3 days in a T25 flask, the culture medium was removed, and cells were washed with 1X PBS. RNA extraction was performed by adding 1 mL of TRIzol reagent (Ambion, Carlsbad, CA, USA) to the flask followed by harvesting of cells using a cell scraper. cDNA was synthesized using the Reverse Transcription Master Premix (ELPIS-BIOTECH, Daejeon, Korea). According to the manufacturer’s instructions, 1.0 μg of mRNA was used as a template to prepare the template RNA Primer Mixture, which was then incubated at 60°C for 1 h and 94°C for 5 mins. Gene expression levels were analyzed using quantitative reverse transcription polymerase chain reaction (RT-qPCR) with Fast qPCR 2x SYBR Green Master Mix (Catalog # EBT-1821, ELPIS-BIOTECH). Amplification was performed over 40 cycles, consisting of denaturation at 95°C for 10 mins, followed by 10 s at 95°C and 20 s at 60°C for each cycle. Target genes included MYH2, MYOG, and DES, with the ACTB gene serving as a housekeeping gene for expression level analysis. Primers used for RT-qPCR are listed in Table 3. The 2−ΔΔCT method was used to quantify mRNA levels [41].
On the third day of differentiation in a T25 flask, the culture medium was removed, and HSC were washed with cold 1X PBS. Cells were then lysed by adding 450 μL of 1X radio-immunoprecipitation assay (RIPA) lysis buffer (Rockland, Gilbertsville, PA, USA) and collected using a cell scraper. Protein concentration in each cell sample was determined using the Bradford assay. Following quantification, cell proteins were mixed with sodium dodecyl sulfate (SDS) sample buffer and heated at 95°C for 5 mins. Proteins were then separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on TGX Precast Gels (Bio-Rad). Following electrophoresis, proteins were transferred to Immun-Blot polyvinylidene fluoride (PVDF) membranes (Bio-Rad). Membranes were blocked with EveryBlot Blocking Buffer (Catalog # 12010020, Bio-Rad) for 5-10 mins and incubated with primary antibodies at 4°C overnight. Primary antibodies used included anti-β-actin (1:1,000, Catalog # PA1-46296, Invitrogen, myosin 4 monoclonal antibody (1:1,000, Catalog # 14-6503-82, Invitrogen), and desmin monoclonal antibody (1:200 [= 5 μg/mL], Catalog # 14-9747-82, Invitrogen). Membranes were washed four times for 5 mins each at room temperature with TBST, a mixture of 10X TBS (Catalog # 1706435, Bio-Rad) and 0.1% Tween 20 (Bio-Rad, USA). After washing, membranes were incubated with Affinity Purified Goat Anti-Mouse (or Rabbit) IgG (H+L) HRP-conjugated antibody (Anti-Mouse [1:5,000, Catalog # 1706516, Bio-Rad], Anti-Rabbit [1:20,000, Catalog # SA002-500, GenDEPOT Katy, TX, USA]) at room temperature for 1 h. Following another set of four washes with TBST for 5 mins each, protein detection was carried out by dispensing 1 mL of Clarity™ Western ECL substrate (Catalog # 170-5061, Bio-Rad) onto each membrane.
To compare proteins of differentiated HSC (CON and D100), each protein sample was analyzed using LC-MS/MS at the Korea Basic Science Institute (KBSI). Analyses of proteins were performed using Bos taurus proteins as reference.
Cells were lysed with RIPA lysis buffer at 4°C for 30 mins and then centrifuged 10,000×g for 10 mins to extract the supernatant containing proteins. Extracted proteins were quantified using a bicinchoninic acid assay kit (Thermo Fisher Scientific). Protein samples were digested using an S-Trap mini spin column (Protifi, Fairport NY, USA). Samples were mixed with 5% SDS in 50 mM TEAB, homogenized, and heated, followed by alkylation with iodoacetamide to a final concentration of 20 mM. Alkylated proteins were acidified using phosphoric acid and then centrifuged at 4,000×g for 30 s. Samples were washed with a solution by mixing methanol with 50 mM TEAB at a 9:1 ratio and digested with trypsin gold (Promega) at a 10:1 (w/w) protein-to-enzyme ratio.
All measurements were conducted at least three times. All statistical analyses were performed by one-way analysis of variance (ANOVA) using SPSS software version 28.0 (SPSS, Chicago, IL, USA). Significant differences among measured values were assessed through Duncan’s Multiple Range Test. Statistical significance was considered when p-value was less than 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001). Although a p < 0.05 was generally used, we have also chosen to use 0.01 (and 0.001) for some of the data to indicate the greater significance of the differences.
RESULTS AND DISCUSSION
Sixteen types of amino acids were analyzed for constituents of drone pupae. Amino acids, which contain both an amino group (−NH2) and a carboxyl group (-COOH), are fundamental building blocks of proteins [42]. Our analysis (Table 4) revealed that glutamic acid was the most abundant amino acid present in drone pupae. Glutamic acid is involved in a greater number of metabolic reactions than other amino acids, functioning as a source of glucose and enhancing the secretion of growth hormones [43]. Furthermore, glutamic acid is utilized in various applications including the development of muscle and other cellular components, enhancement of immune functions, anticancer activities, flavor enhancement, and as a constituent in culture media [43]. Following glutamic acid, aspartic acid was the second most abundant amino acid. It is involved in energy metabolism, fatigue resistance, and enhancement of immune function [44]. Essential amino acids such as threonine, valine, methionine, isoleucine, leucine, phenylalanine, lysine, and histidine were also detected in drone pupae. Among these, leucine, isoleucine, and valine belong to branched-chain amino acids (BCAA) primarily used as muscle energy sources [45]. Among them, leucine has been shown to activate mTORC1 and contribute to the prevention of muscle atrophy [46]. Beyond BCAA, various amino acids can regulate muscle protein breakdown and synthesis. They are used to mitigate muscle atrophy associated with nutritional deficiency and aging [46]. Furthermore, glycine can protect against muscle wasting in C2C12 myoblasts and lysine can attenuate sarcopenia by inhibiting protein degradation in a mouse model of age-related sarcopenia [47,48].
The analysis of vitamins in drone pupae revealed the presence of water-soluble vitamins (vit) such as vit B1 (1.50 mg/100 g), vit B6 (28.76 mg/100 g), and vit C (19.92 mg/100 g) and fat-soluble vit E (0.17 mg/100 g) (Table 5). Vit B12 was not detected. Vit B1 is essential for proper cellular function, contributing to energy production in mitochondria, antioxidant, anti-inflammatory, and antitumor activities. It is also important for maintaining neural function [49]. Vit B6 is crucial for cellular metabolism. It exhibits antioxidant properties, including an ability to suppress reactive oxygen species (ROS) [50,51]. Furthermore, it contributes to the suppression of muscle loss, satellite cell proliferation, reduction in cell death, and promotion of muscle growth [52,53]. Vit C is known for its potent antioxidant and anti-inflammatory effects, which can stimulate skeletal muscle growth and regeneration, potentially improving muscle atrophy [54,55]. Sawczuk et al. [56] have shown results similar to those reported for vitamin C abundance in drone pupae. Vit E also possesses antioxidant properties. It can safeguard cell membranes from oxidative damage induced by ROS [57,58]. According to Chung et al. [59], vitamin E can aid in myoblast proliferation, differentiation, mitochondrial efficiency, and muscle mass.
| Traits (mg/100 g) | Contents |
|---|---|
| Vit B1 | 1.50 ± 0.08 |
| Vit B6 | 28.76 ± 0.65 |
| Vit B12 | ND |
| Vit C | 19.92 ± 0.03 |
| Vit E | 0.17 ± 0.01 |
The analysis of minerals in drone pupae revealed that K and P were the most abundant minerals in drone pupae, with concentrations of 10,218.32 ± 62.17 mg/kg and 6,681.65 ± 64.50 mg/kg, respectively (Table 6). Other minerals such as Mg, Na, Ca, Fe, Zn, and Cu were also detected in a descending order of abundance, while Mn was not detected. K plays a critical role in maintaining muscle and nerve function and P is essential for maintaining and repairing all cells and tissues [60,61]. Mg is pivotal in energy metabolism, muscle contraction, and muscle relaxation. Its depletion can lead to increased oxidative stress and structural damage to muscle cells through impaired intracellular calcium homeostasis [62,63]. Furthermore, Mg can serve as an antioxidant to protect mitochondria from free radical damage [64]. Ca is a crucial ion that can trigger muscle contraction. It plays important roles in cellular metabolism, bone formation, and protein synthesis [65,66].
| Traits (mg/kg) | Contents |
|---|---|
| Ca | 370.09 ± 3.42 |
| Cu | 14.81 ± 0.90 |
| Fe | 78.84 ± 4.08 |
| K | 10,218.32 ± 62.17 |
| Mg | 692.53 ± 4.32 |
| Mn | ND |
| Na | 395.24 ± 3.06 |
| Zn | 51.20 ± 0.25 |
| P | 6,681.65 ± 64.50 |
ROS are oxygen-centered free radicals, including superoxide (O2−), hydroxyl radicals (HO·), and hydrogen peroxide (H2O2) [67]. The formation of ROS is crucial for maintaining cellular homeostasis. It occurs due to metabolism, normal respiration, and stress associated with diseases [68,69]. It is involved in a variety of cellular processes, from apoptosis and necrosis to proliferation and carcinogenesis [70]. However, levels of ROS above physiological concentrations can induce oxidative stress, which antioxidants can mitigate [71]. One method to evaluate antioxidant capacity involves assessing protective effects against oxidative stress induced by H2O2 [72].
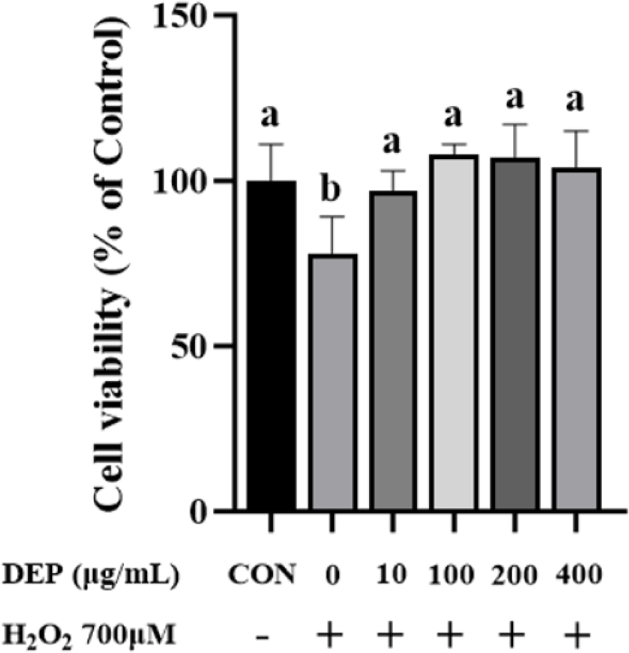
Fig. 2 presents result of an experiment examining the antioxidative power of DEP by measuring cell viability in response to H2O2-mediated damage. The survival rate of HSC treated with 700 μM H2O2 showed a significant decrease compared to that of CON (p < 0.05). The survival rate of H2O2-treated HSC was restored to 100% of CON level after adding DEP at 100, 200, or 400 μg/mL (DEP 100 μg/mL: 108%, DEP 200 μg/mL: 107%, DEP 400 μg/mL: 104%). Among DEP treatments, the 100 μg/mL concentration resulted in the highest numerical cell survival rate. This suggests that the presence of phenolic compounds within drone pupae may confer antioxidant properties [73]. Lee et al. [74] have reported that the cytotoxicity induced by H2O2 can be mitigated and cell viability can be restored when mouse muscle cells (C2C12) are treated with Protaetia brevitarsis larvae protein extract. This result suggests that DEP can protect HSC from oxidative stress damage induced by H2O2.
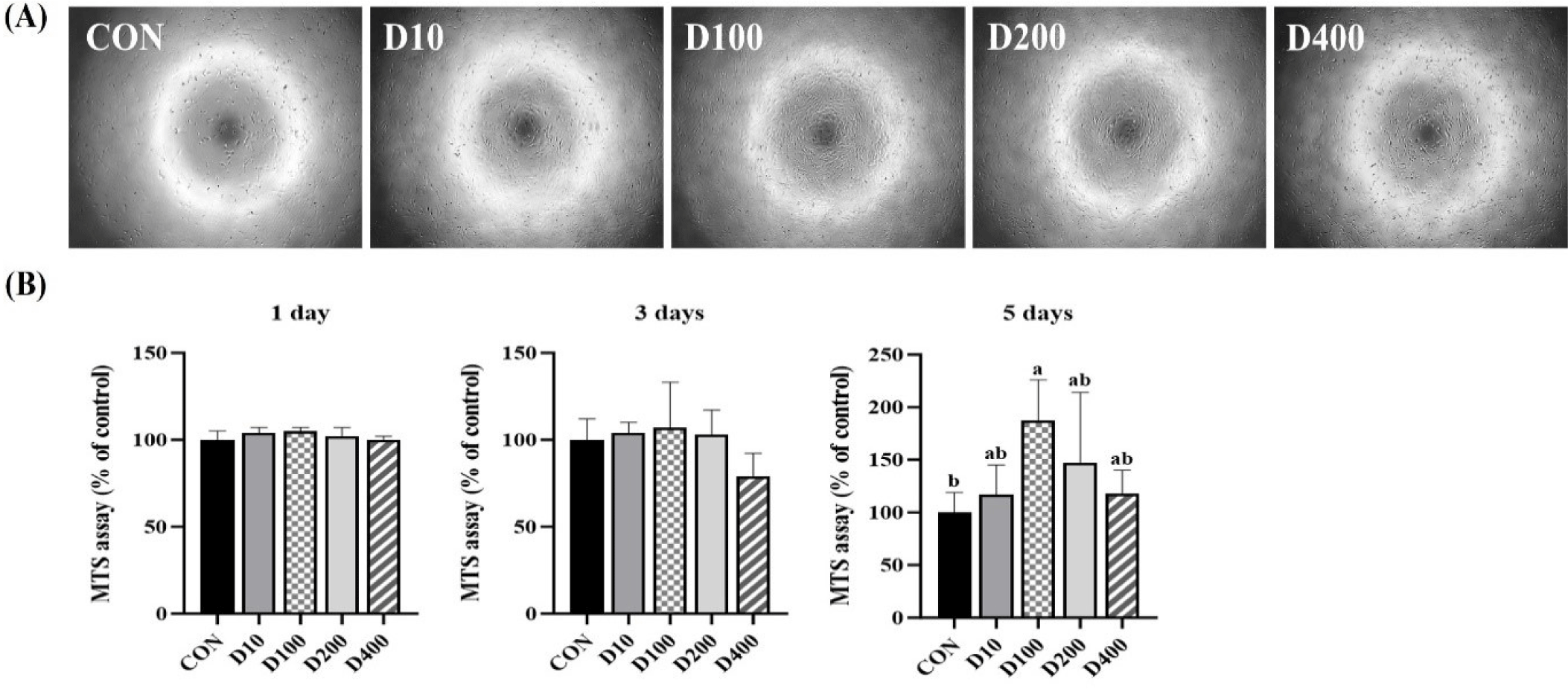
An MTS assay was conducted to determine the concentration of DEP at which HSC exhibited the highest viability. HSC were cultured in GM supplemented with DEP at concentrations of 10, 100, 200, and 400 µg/mL for five days. According to MTS assay results (Fig. 3), there was no significant difference in cell viability between treatments on day 1 or day 3. However, after 5 days, the D100 group treated with 100 µg/mL DEP showed significantly higher cell viability than the CON group (p < 0.05), while DEP-treated groups showed no significant differences in cell viability. The addition of DEP led to enhanced cell viability. This result is attributed to antioxidant properties of DEP, as evidenced by results of H2O2 scavenging activity assay (Fig. 2). This suggests that DEP may mitigate oxidative stress and enhance cell viability, self-renewal capacity, and differentiation potential [75]. Drowley et al. [76] have demonstrated that elevating antioxidant levels can enhance the proliferation of muscle-derived stem cells and improve their tissue regeneration capabilities. Furthermore, amino acids, minerals, and vitamins present in drone pupae (Tables 4, 5 and 6) could also influence cell viability. Based on the results of the proliferation culture, we also investigated effect of DEP on differentiation capacity of HSC after treating cells with 100 μg/mL of DEP and CON.
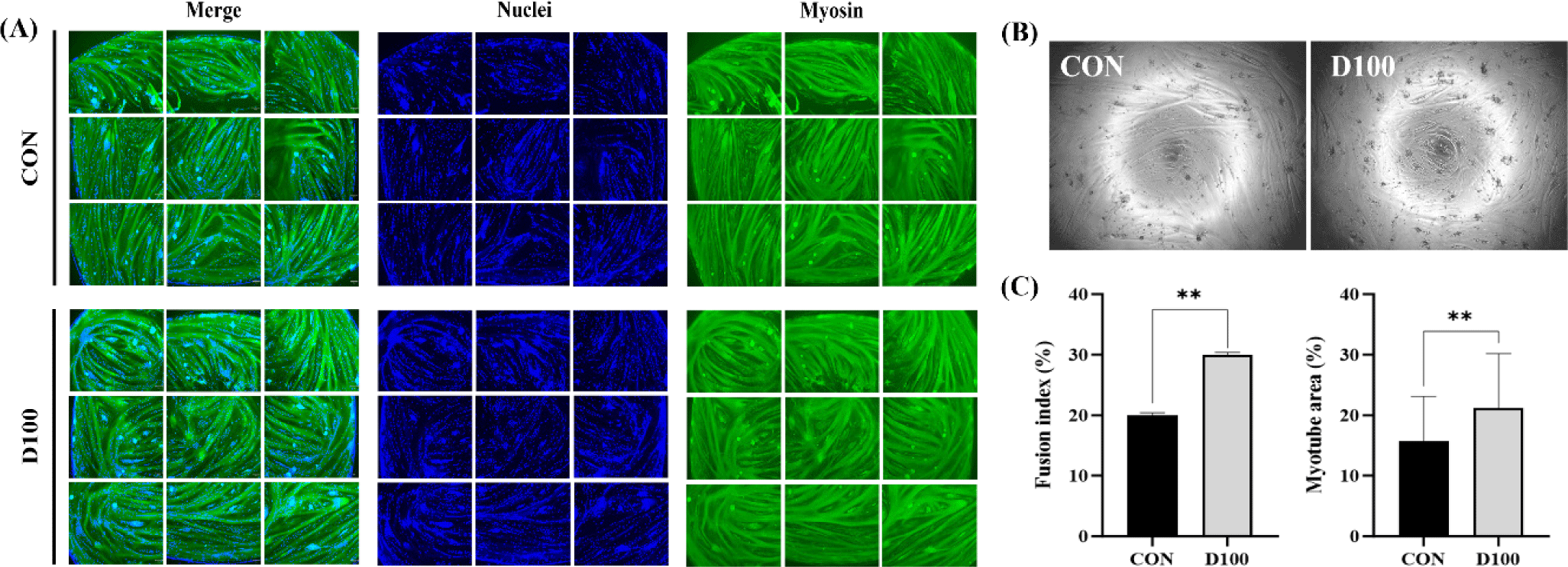
Immunofluorescence staining was performed to analyze nuclear distribution and myosin protein expression in HSC differentiated for four days in the presence of DEP at a concentration of 100 μg/mL (Fig. 4A). Fig. 4B displays an image of differentiated HSC for 3 days before immunofluorescence staining. Myosin protein expression facilitated greater myotube formation in D100 than in CON. Images of stained cells were analyzed to measure muscle cell differentiation characteristics such as the fusion index and myotube area (Fig. 4C). The Fusion Index, a primary marker for quantifying satellite cell differentiation, is defined as the percentage of nuclei per myotube relative to the total number of nuclei in the sample [77]. Both the Fusion Index and myotube area were significantly higher in D100 than in CON (p < 0.01). Kang et al. [78] have reported that treating C2C12 cells with silkworm (Bombyx mori) protein hydrolysate at concentrations above 100 μg/mL can increase the fusion index. Based on this result, it is believed that DEP can promote myotube formation in HSC.
To analyze the impact of DEP on the differentiation capacity of HSC, we assessed expression levels of genes and proteins related to myogenesis using RT-qPCR and Western blot, respectively (Fig. 5). Myosin, a major structural protein in muscle cells, is abundant in skeletal muscles. It plays a crucial role in muscle contraction and relaxation [79]. It serves as a cellular differentiation marker, being a primary component of the myosin filament in myofibrils [80]. Myogenin is expressed during the differentiation of myoblasts into multinucleated myotubes, a process also confirmed in the differentiation of bovine satellite cells [81,82]. Desmin located in actin filaments is a principal protein of the muscle cell cytoskeleton. It is specific to muscle cells, playing a critical role in normal muscle function [83]. It is expressed at the start and completion of muscle formation. It is a key marker of muscle differentiation, accumulating during an in vitro myogenesis process [84]. As cells fuse into myotubes, the expression of structural proteins such as desmin increases [85]. For this reason, these three genes were selected and subjected to experiments.
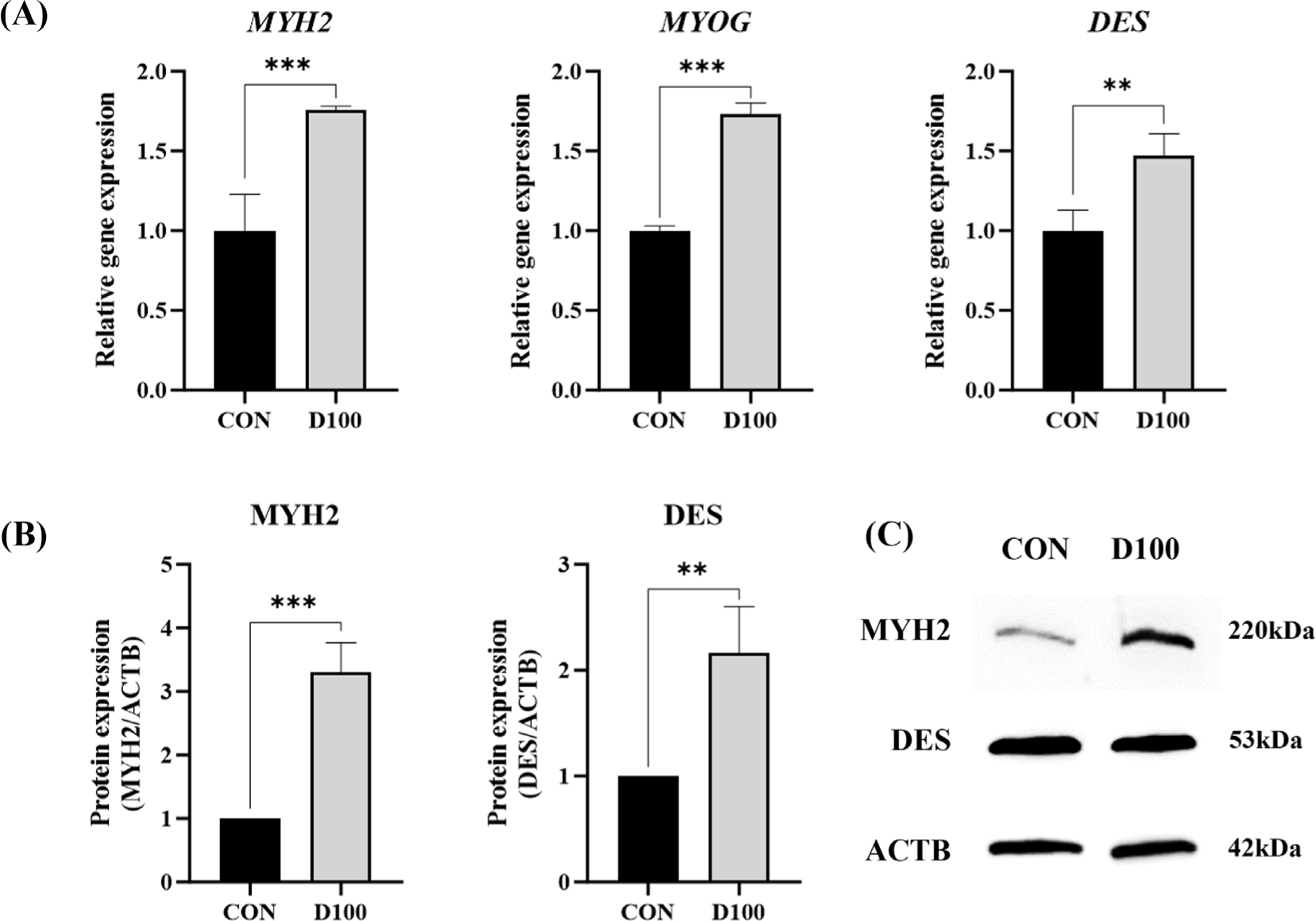
RT-qPCR was employed to compare gene expression levels of MYH2, MYOG, and DES in HSC differentiated for 3 days in the presence of DEP at a concentration of 100 µg/mL (Fig. 5A). Gene expression levels of MYH2 and MYOG in the D100 group were significantly higher than those in CON (p < 0.001). Lee et al. [86] have reported that the expression of myogenin is increased by 1.34-fold in HSCs treated with crude polysaccharides from Ecklonia cava hydrolysate. DES gene expression is also significantly increased in the D100 group compared to that in CON (p < 0.01). Ciecierska et al. [87] have reported that the high expression of desmin in muscle cells from various beef breeds is associated with muscle cell differentiation and myoblast fusion according to functional analyses. Choi et al. [88] have demonstrated an increase in myogenin gene expression in C2C12 myotubes treated with ethanol extract of Tenebrio molitor larvae compared to controls. Findings of this study suggest that DEP can enhance HSC differentiation by upregulating the expression of genes associated with myogenesis.
Fig. 5B shows results of a western blot analysis comparing protein expression levels of MYH2 and DES in HSC differentiated for 3 days at a concentration of 100 µg/mL DEP. The intensity of western blot band was used to measure protein expression of MYH2 and DES (Fig. 5C). Expression levels of MYH2 and DES proteins were significantly higher in D100 than in CON (p < 0.001 and p < 0.01, respectively). Rønning et al. [82] have reported that desmin protein expression is significantly increased in differentiated primary bovine skeletal muscle cells. Kang et al. [78] have demonstrated that treatment with silkworm protein hydrolysate can increase MyHC protein expression in differentiated C2C12 cells. Based on this result, it is believed that DEP can promote myotube formation in HSC.
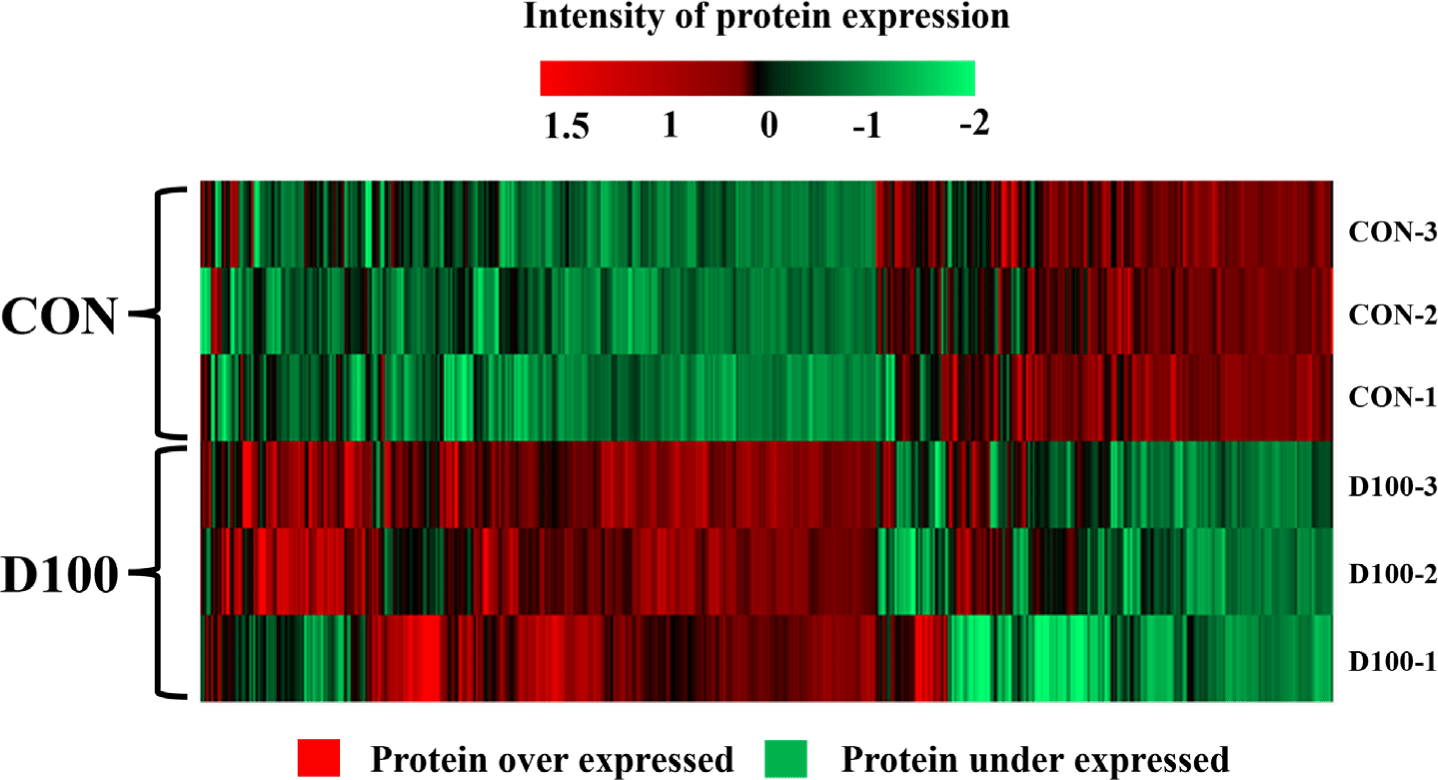
Proteome analysis was conducted to compare proteins of HSC differentiated for 4 days in the presence of DEP at a concentration of 100 μg/mL. Protein expression intensity is displayed using a heatmap (Fig. 6), where red indicates upregulated protein expression and green indicates downregulated protein expression. D100 comprised thousands of different protein types, with a total of 1,705 proteins measured. Of these, 1,057 proteins were expressed at more than twice the expression intensity of CON, indicating upregulation, while 627 proteins were expressed at less than half the intensity of CON, indicating downregulation. Proteins upregulated in D100, such as myosin, IL18, MYO1D, and LIM domain proteins, are involved in functions such as activating muscle cell metabolism, providing antioxidant effects, and promoting muscle cell differentiation [89–91]. It appears that proteins upregulated by DEP treatment can affect the differentiation of HSC.
CONCLUSION
In this study, we investigated effects of DEP on proliferation and differentiation of HSC. Initially, we analyzed amino acids, vitamins, minerals, and antioxidant properties to characterize drone pupae. As a result, amino acids such as glutamic acid and BCAAs, vitamins such as vitamin C, and minerals were detected. Furthermore, DEP demonstrated antioxidant properties by enhancing HSC survival rate, which had been diminished by H2O2 exposure.
These properties and effects of DEP might have contributed to enhanced proliferation and differentiation abilities of HSC. Results of this study revealed that cell activity of HSC treated with DEP at various concentrations was higher than that of CON, with the group treated with DEP at 100 μg/mL showing the highest activity. Furthermore, treatment with DEP at 100 μg/mL enhanced expression levels of genes and proteins related to muscle formation. In differentiation culture of DEP-treated HSC, the proteome analysis during muscle formation was upregulated proteins including myosin and IL18 proteins, which are linked to energy production, promotion of differentiation, and antioxidant activities.
The positive effect of DEP in enhancing the proliferation and differentiation abilities of HSC might be attributed to the influence of various components present in DEP. This demonstrates its potential as a functional material for enhancing HSC proliferation and differentiation in future production of cultivated meat. Further research is required to build on this study and gain a more detailed understanding of how DEP enhances HSC proliferation and differentiation.