
Bovine digital dermatitis (BDD) is a highly contagious disease affecting the hoof of cattle, characterized by the development of ulcerative lesions and chronic hyperkeratosis [1]. The disease causes significant lameness, resulting in substantial losses for both dairy and beef producers due to decreased efficiency in milk production and average daily gain [2]. BDD mainly affects the interdigital area of intensively managed cattle, especially high-yielding dairy cows, and spreads through direct contact and contaminated environments [3]. BDD is a polybacterial disease, and previous research showed that multiple Treponema species are involved, with T. medium, T. pedis, and T. phagedenis considered as the core species responsible for the development and progression of the disease [4]. The present study focused on the whole genome sequencing (WGS) of T. pedis GNW45 isolated from BDD in Korea. This analysis could provide insights into the virulence factors and pathogenic mechanisms underlying the disease.
T. pedis GNW45 was isolated from a Holstein-Friesian cow suffering from BDD in a farm in Hwaseong, Gyeonggi province. Under anaerobic conditions, GNW45 was isolated and purified following the protocol described in a previous study [5]. DNA of GNW45 was purified and was analyzed for quality inspection and WGS. Sequencing and assembly were performed as previously described [6]. In brief, a library for long-read sequencing was prepared using the PacBio 20 kb SMRTbellTM kit for the PacBio RSII platform (Pacific Biosciences, Menlo Park, CA, USA), generating 188,880 total subreads. Concurrently, a short-read library was prepared using the TruSeqNano DNA Kit for the Illumina HiSeqXTen platform, which produced 8,990,700 filtered short reads. De-novo assembly was performed by mapping the PacBio RSII single-pass reads to seed reads using the Hierarchical Genome Assembly Process. A single circular contig was generated after utilizing the PacBio long reads. To improve accuracy and quality, sequence compensation and error correction were performed by aligning the Illumina reads to the pre-assembled genome using Pilon v1.21. Annotation was performed using Prokka v1.12b. Functional annotation of protein-coding sequences (CDS) was carried out using EggNOG v5.0 and mapped in circular presentation using Proksee server.
The genome of T. pedis GNW45 is 3,077,465 bp long, and is composed of 2,783 genes. The sequencing and annotation statistics is presented in Table 1, and the circular representation of the genome properties and the summary of functional annotation is shown in Figs. 1A and 1B, respectively. The genus Treponema is composed of a diverse number of species from a variety of niches, and can play as either commensal or pathogen, or both [7]. Other studies have isolated T. pedis from infections in animals other than BDD, like necrotic skin ulcer and gingiva in pigs, and hoof canker in horses, demonstrating that it could thrive in an environment with a diverse microbial community [7], contributing to progression of polybacterial diseases. However, the role of this bacterium in the complex bacterial community of BDD is still not clear, hence we highlighted in this report some of the important properties of this bacterium that could potentially contribute to its pathogenicity.
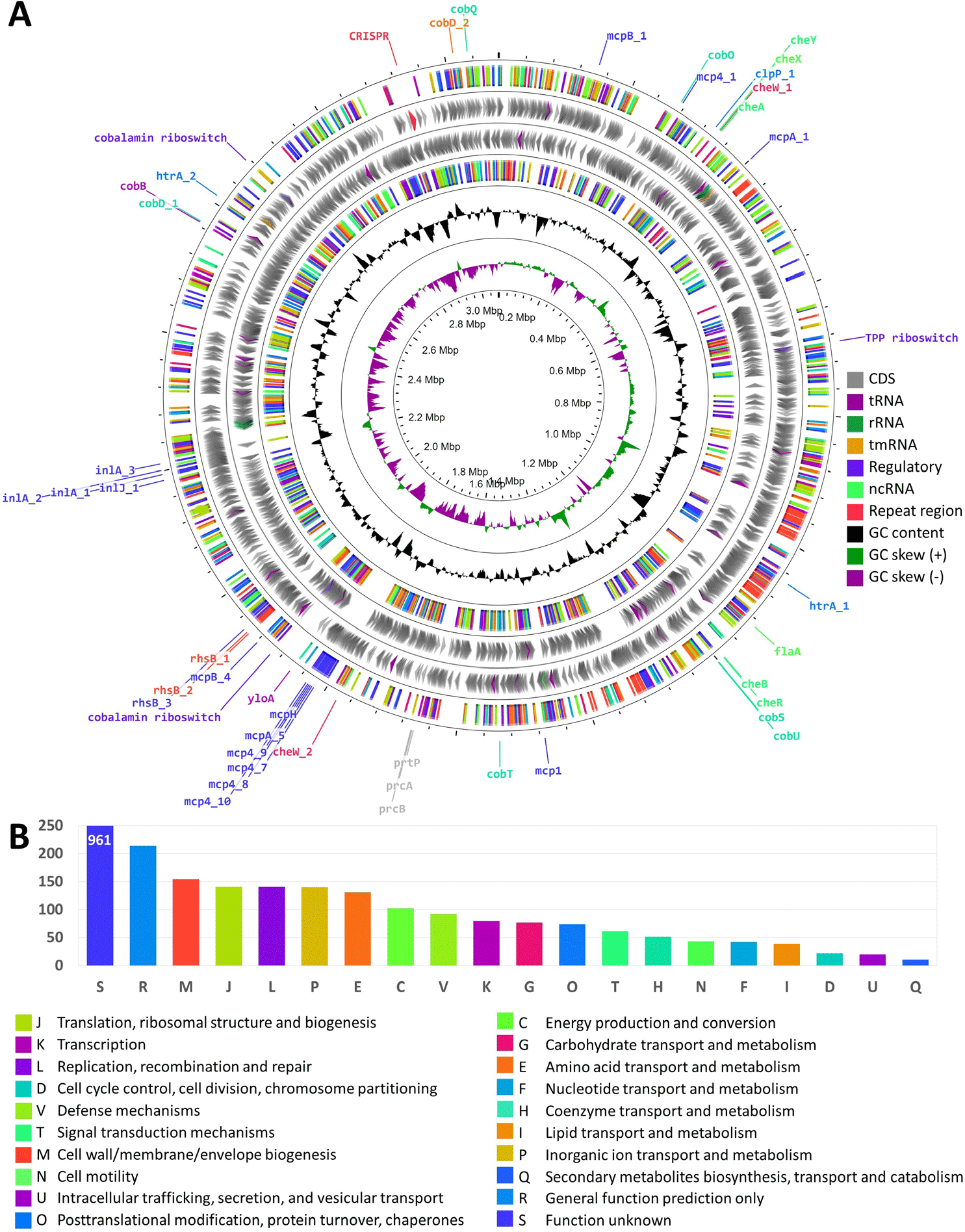
The helical morphology of T. pedis is regulated by multiple flagellar-motility associated proteins, enabling its chemotactic response to specific chemical signals through various chemotaxis proteins. These mechanisms involve 58 genes for flagellar biosynthesis, 97 genes for chemotaxis, and four genes for motility. Furthermore, the presence of chemoreceptors, such as methyl-accepting chemotaxis proteins, assists in bacterial adherence to the host, in conjunction with motility. Additionally, genes for fibronectin-binding protein (yloA), dentilisin (prtP, prcA, prcB), major surface protein (Msp), serine proteases (htrA, clpP), and internalins (inlA, inlJ), have been identified to have roles in adhesion, host invasion, and immune evasion.
T. pedis has genetic adaptations for metabolic competition, including 59 Rearrangement hotspots and YD-repeat proteins that inhibit neighboring cell growth, enabling competitive exclusion [8]. Beyond competitive adaptations, the BDD microbiome possesses genetic elements that may facilitate cooperative metabolic interactions with other microbes. Regulatory riboswitches and genes involved in Vitamin B12 and porphyrin biosynthesis (cob’s) and transport have been identified. Porphyrin has been demonstrated to induce a proinflammatory host response in other skin infections [9]. Furthermore, the presence of a Type 2C clustered regularly interspaced palindromic repeats (CRISPR)-Cas system has been detected, with the repeat region encompassing a total of 141 spacers, revealing its previous exposure to foreign genetic elements which may reflect the strain’s environmental history and potential virulence [10].
The WGS of T. pedis GNW45 from BDD revealed genes for motility and chemotaxis, and competitive and cooperative interactions. It also possesses host adhesion-related genes, a CRISPR-Cas system, several proteases, and porphyrin biosynthesis genes, suggesting its ability to adhere, invade, and modulate the inflammatory immune response of the host. These findings enhance our understanding of pathogenicity of T. pedis in BDD and its complex interactions within the microbial ecosystem, highlighting its virulence mechanisms for infection and colonization.