INTRODUCTION
Interactions between the microbiome and diet affect microbial colonization of the gastrointestinal tract in pigs. Specifically, dietary fiber supplementation in pigs has been associated with both negative and positive effects. A high fiber diet increases the expression level of Lactobacillus spp. in the digestive tract and enhances the volatile fatty acids (VFAs) production in the hindgut of weaned piglets [1]. VFA production is essential for regulating metabolism and contributes to animal health [2]. Insoluble fiber content reportedly induces changes in the gut microbiota, specifically by lessening the Firmicutes:Bacteroidetes (F/B) ratio and the expression level of Lactobacillus spp., which leads to increased susceptibility of pigs to colitis [3]. Additionally, different dietary protein sources affect the microbial composition of pigs. For example, cottonseed meal-based diets have been shown to increase the expression of Lactobacillus spp., which may improve gut health. In contrast, a fishmeal-based diet increases the abundance of Escherichia spp. and Shigella spp., which in turn increases the likelihood of diarrhea [4].
Sasa quelpaertensis Nakai (SQ) is a perennial bamboo species belonging to the grass family Poaceae, and is known for its high protein and fiber content. SQ is widespread, particularly on Mount Halla, Jeju Island, Korea. A few researchers have assessed the influences of SQ as a potential ingredient in functional materials. SQ extract (SQE) has been shown to exert various health benefits, exhibiting antidiabetic, antioxidative, anti-inflammatory, and antiviral effects [5–7]. More recently, Lee et al. reported that, compared with rice straw, SQ has superior feed value as a roughage source for Hanwoo cattle when fed as part of a total mixed ration [8].
Despite the positive effects of SQ highlighted in previous research, the influences of SQ supplementation on the gut microbial communities of livestock remain unknown. Furthermore, the effects of SQ supplementation on important phenotypes related to livestock productivity, such as growth performance and carcass traits, have not been investigated. Therefore, in the present study, using SQE supplementation and control groups, and changes in the microbial composition and diversity in pigs were assessed using 16S rRNA amplicon sequencing. The influences of SQE supplementation on average daily gain (ADG) and backfat thickness (BF) were also assessed after slaughter. This study is, to our knowledge, the first to assess the influences of SQ supplementation on intestinal bacteria in pigs. We believe that these research findings will be helpful in demonstrating the potential of SQ as a feed resource and its ability to enhance animal productivity.
MATERIALS AND METHODS
All experiments involving animals presented in this research were approved by the Institutional Animal Care and Use Committee (IACUC) of the National Institute of Animal Science (NIAS) (approval number: NIAS20212189). A total of 14 Landrace pigs were used in the study, with seven pigs (five males and two females) in the control group and seven pigs (two males and five females) in the SQE supplementation group. The 14 Landrace pigs were raised in a standardized environment at the Subtropical Livestock Research Institute (Jeju, Korea). The age of the pigs at the start of the experiment ranged from 151 to 160 d, with an average age of 154.8 d. The average ± standard deviation of body weight of the control group and SQE supplementation group were 80.57 ± 10.79 kg and 80.00 ± 11.28 kg, respectively. The experiment was conducted in the same rearing place for 41 d, wherein the control group was fed a commercial formula at a rate of 2.25 kg per pig twice a day, for a total of 4.5 kg per day. The SQE supplementation group received at same frequency and amount as the control group, with an additional 450 ml of SQE added to the commercial formula per feeding by top dressing. The chemical composition of the commercial formula and SQE are listed in Table 1.
The Jeju Plant Resources Institute (Jeju, Korea) obtained permission from Jeju Island and collected SQ on Mount Halla. The collected SQ was washed and dried at 60°C for 8–14 h. SQE was prepared as a mixture of dried SQ and water in a 1:20 ratio, which was then subjected to high-pressure extraction at 10060°C for 3 h. To calculate ADG, the initial body weight and the final body weight after 41 d of the experiment were measured in 14 Landrace pigs, and the weight differences were divided by the experimental period of 41 d. The 14 Landrace pigs were slaughtered within eight days after the end of the experiment to measure the BF in Jeju Livestock Cooperative (Jeju, Korea). The BF information of 14 Landrace pigs was acquired from the Korea Institute for Animal Products Quality Evaluation (https://www.ekape.or.kr).
After the experiment was completed, fecal samples were acquired from the 14 pigs and stored at −7060°C. Genomic DNA was extracted from a fecal sample (250 mg) using the QIAamp PowerFecal Pro DNA kit (Qiagen, Hilden, Germany). The quantity and quality of the extracted genomic DNA were measured using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The V3-V4 hypervariable segments of the 16S ribosomal RNA gene were amplified from the fecal genomic DNA using the universal primer sets Bakt_341F (CCTACGGGNGGCWGCAG) and Bakt_805R (GACTACHVGGGTATCTAATCC). The input DNA 2 ng was polymerase chain reaction (PCR) amplified with 5x reaction buffer, 1 mM of dNTP mix, 500 nM each of the universal F/R PCR primer, and Herculase II fusion DNA polymerase (Agilent Technologies, Santa Clara, CA, USA). Following purification, the 2 μL of 1st PCR product was PCR amplified for final library construction containing the index using Nextera XT Indexed Primer. The cycle condition for 2nd PCR was same as the 1st PCR condition except for 10 cycles. The PCR product was purified with AMPure beads. The final purified product is then quantified using quantitative PCR (qPCR) according to the qPCR Quantification Protocol Guide (KAPA Library Quantification kits for Illumina Sequencing platforms) and qualified using the TapeStation D1000 ScreenTape (Agilent Technologies, Waldbronn, Germany). The sequencing of amplified products was conducted using an Illumina MiSeq platform (Illumina, San Diego, CA, USA).
The microbiome taxonomic profiling (MTP) of EzBioCloud (ChunLab, Seoul, Korea) was utilized to perform microbial classification. Briefly, the sequencing reads were processed as follows: 1) paired reads were merged into a single read; 2) forward and reverse primer sequences were trimmed; 3) low-quality (< Q25) reads were filtered; 4) non-redundant reads were denoised and extracted; 5) chimeric reads were detected and removed; and 6) operational taxonomic units (OTUs) with similarity greater than 97% were selected. Microbial taxa were classified using the EzBioCloud 16S rRNA database (version PKSSU4.0) [9]. The taxonomic composition was normalized using the copy number of the 16S rRNA genes. Good’s coverage for sequencing depth and diversity indices (Chao1, Shannon, and Simpson) of the microbial communities were estimated to identify species richness and diversity. Principal coordinate analysis (PCoA) method was employed to measure beta diversity using UniFrac distance matrices, including unclassified OTUs at the species level.
The Wilcoxon rank-sum test was employed to assess the difference in ADG, BF, and microbial diversity between groups. The results were considered statistically significant at p < 0.05. We used linear discriminant analysis effect size (LEfSe) method to measure the effect size of taxa, and defined taxa with a linear discriminant analysis (LDA) score > 4 and p < 0.05 as microbiota with differential expression between groups [10].
RESULTS & DISCUSSION
The total number of bases (bp), reads, guanine cytosine (GC) (%), Q20 (%), and Q30 (%) were calculated to estimate the quality of the 16S rRNA amplicon sequencing (Table 2). To investigate how SQE supplementation affects the gut microbial environment, the relative proportion of the taxa in both groups was compared at the different taxonomic levels (Figs. 1A, 1B, 1C and 1D). Firmicutes, Bacteroidetes, Spirochaetes, and Actinobacteria were identified as the most common phyla in both the control and SQE groups (Fig. 1A). LEfSe results indicated that the expression level of Firmicutes (LDA score = 4.69, p = 0.008) and Actinobacteria (LDA score = 4.44, p = 0.018) increased substantially, whereas Bacteroidetes (LDA score = 4.49, p = 0.006) and Spirochaetes (LDA score = 4.61, p = 0.025) decreased in the SQE group (Fig. 1E). The proportion of Firmicutes was 76.48% in the SQE supplementation group and 67.28% in the control group, while that of Bacteroidetes was 8.32% and 14.26%, respectively. The F/B ratio was increased in the SQE group (9.19) relative to the control group (4.71). Higher F/B ratios have been linked to increased energy absorption and accumulation in humans [11]. Additionally, obese pigs reportedly show elevated F/B ratios in their gut microbiota [12]. There were no notable differences observed in ADG between groups (p = 0.52), the ADG showed a tendency to increase in the SQE group (Fig. 2A). These results suggest that the increase in F/B ratios due to SQE supplementation may affect host energy metabolism and body weight gain. The SQE group (3.79%) exhibited a lower expression level of Spirochaetes in comparison to the control group (10.62%). Spirochaetes are intestinal pathogens that cause various diseases, including mucohemorrhagic colitis, typhlitis, and cholera [13]. SQE has been shown to be effective in reducing inflammation in intestinal epithelial cells [14], and recently, a correlation has been reported between an elevated expression of Spirochaetes and the occurrence of African Swine Fever [15]. These results suggest that SQE supplementation can potentially suppress inflammation and prevent disease by reducing the expression level of Spirochaetes. The abundance of Actinobacteria was elevated in the SQE group (11.28%) versus the control group (5.22%). Actinobacteria are actively involved in preserving gut homeostasis and development of the immune system [16]. Based on these results, maintaining the balance and stability of the microbial environment within the immune system can protect pigs from disease.
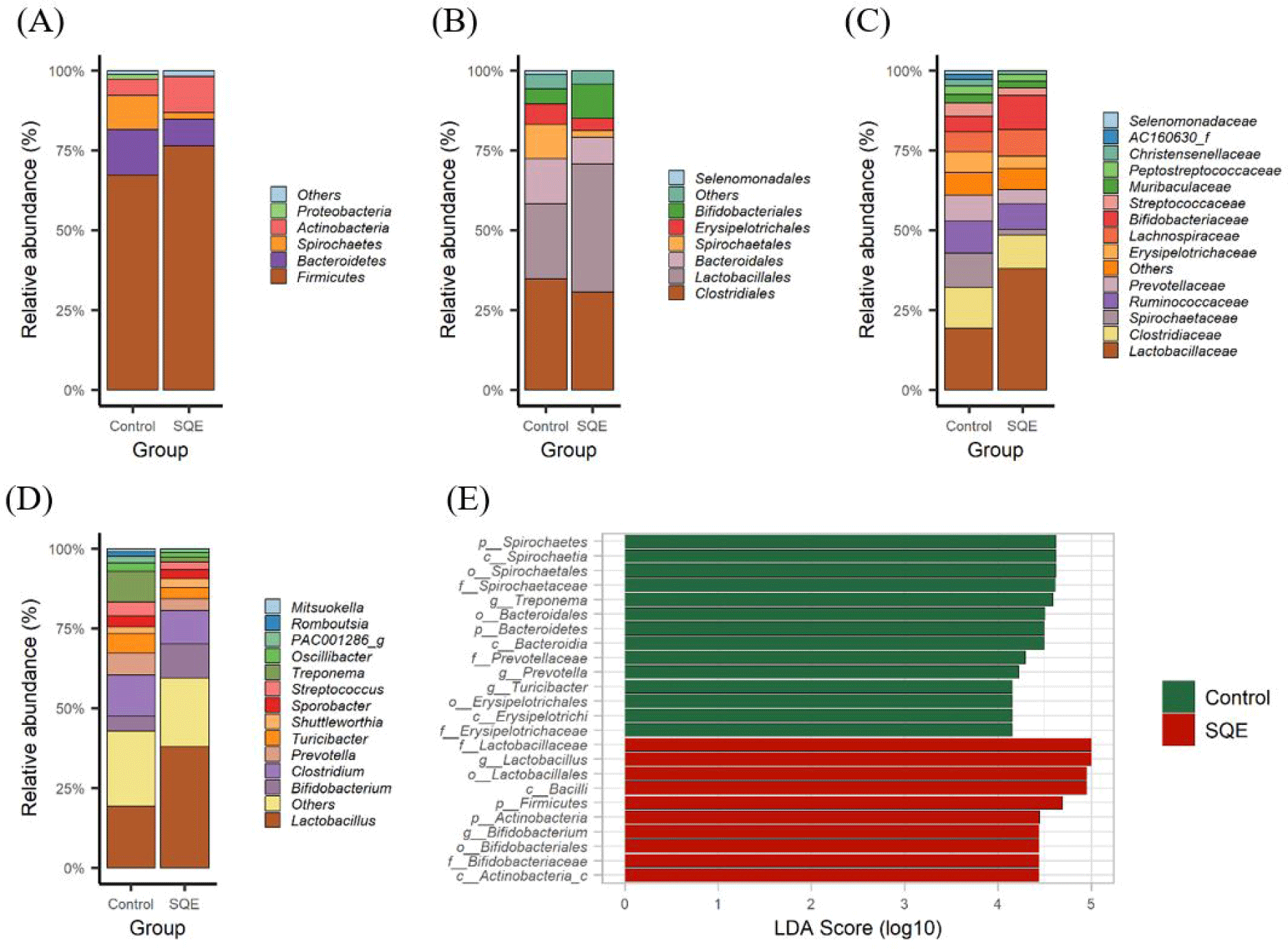
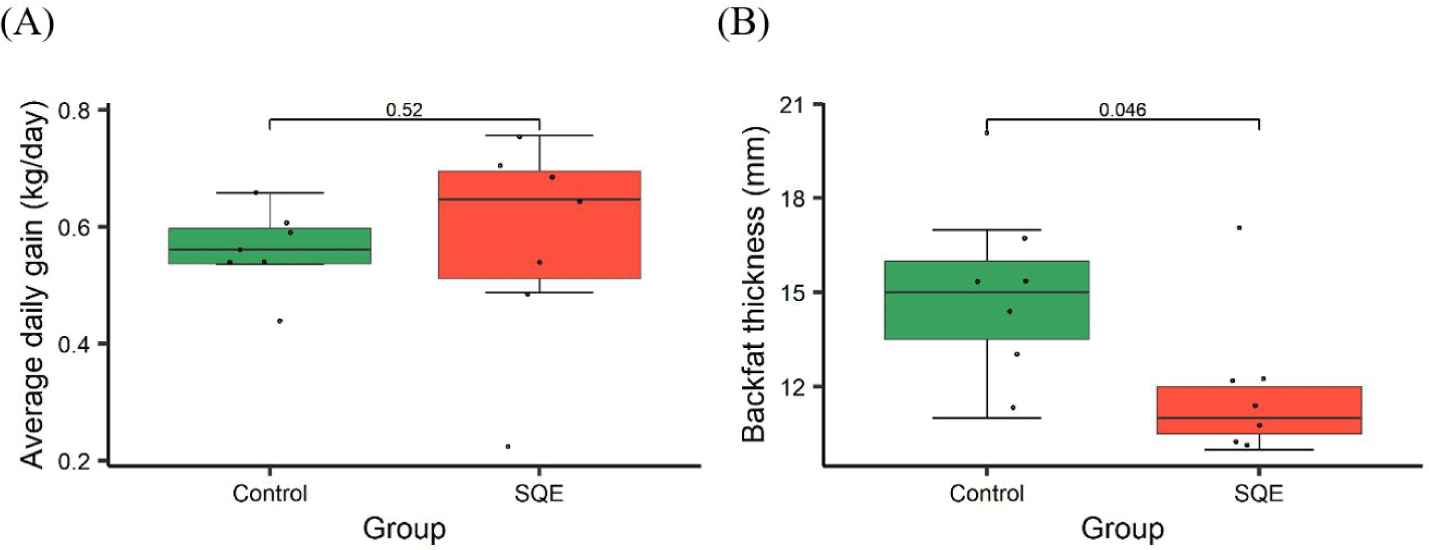
The proportion of Bifidobacterium (LDA score = 4.44, p = 0.018) and Lactobacillus (LDA score = 5.00, p = 0.004) genera was higher in the SQE group, whereas that of Treponema (LDA score = 4.58, p = 0.006), Prevotella (LDA score = 4.21, p = 0.035), and Turicibacter (LDA score = 4.15, p = 0.025) decreased in the SQE group (Fig. 1E). Lactic acid bacteria (LAB), including Lactobacillus and Bifidobacterium regulate the intestinal environment in pigs. LAB can inhibit or eliminate pathogenic agents in the digestive tract, improving the microbiome balance and preserving the intestinal barrier [17]. Interestingly, pigs with low BF have a higher abundance of Lactobacillus reuteri in the gut [18]. Furthermore, Lactobacillus spp. were associated with a decrease in fat mass in mice provided with a high-fat diet [19]. We observed a significant decrease in BF in the SQE group as opposed to the control group (p < 0.05) (Fig. 2B). A study has reported a positive connection between a high abundance of Prevotella copri and elevated levels of serum metabolites associated with obesity [20]. SQE supplementation can reduce the weight of fat tissue in the obesity mouse model and regulate abundances of key proteins participating in fat metabolism in rats provided with a high-fructose diet [21,22]. Our results suggest that SQE supplementation in pigs can increase the expression level of Lactobacillus and decrease the expression level of Prevotella, resulting in a reduction in BF.
Diet is a major factor that affects the gut microbial composition, and environmental conditions and age also affect the composition of the microbiome. In this study, we were limited to raising 14 pigs in the same rearing space during the experimental period in order to minimize the alterations of microbiome caused by environmental conditions. To minimize the changes in the microbiome caused by age differences, we excluded pigs that were more than two weeks apart in age from the experimental group and recruited pigs that were born around the same time. In this study, we performed a comparative analysis of the gut microbiome compositions in a small number of pigs. However, to increase the statistical power of the LEfSe method, we increased the LDA score threshold from the typical value of 2 to 4. A higher LDA score means that the gut microbiome is more likely to be present in one group at a higher relative abundance than in the other group. Although the sample size is small, we believe that results of microbial changes due to the additional feeding of SQE are acceptable because the LDA score threshold was increased. In addition, further studies on a larger number of pigs are needed to investigate the effects of SQE feeding on various growth performances such as feed intake and feed efficiency.
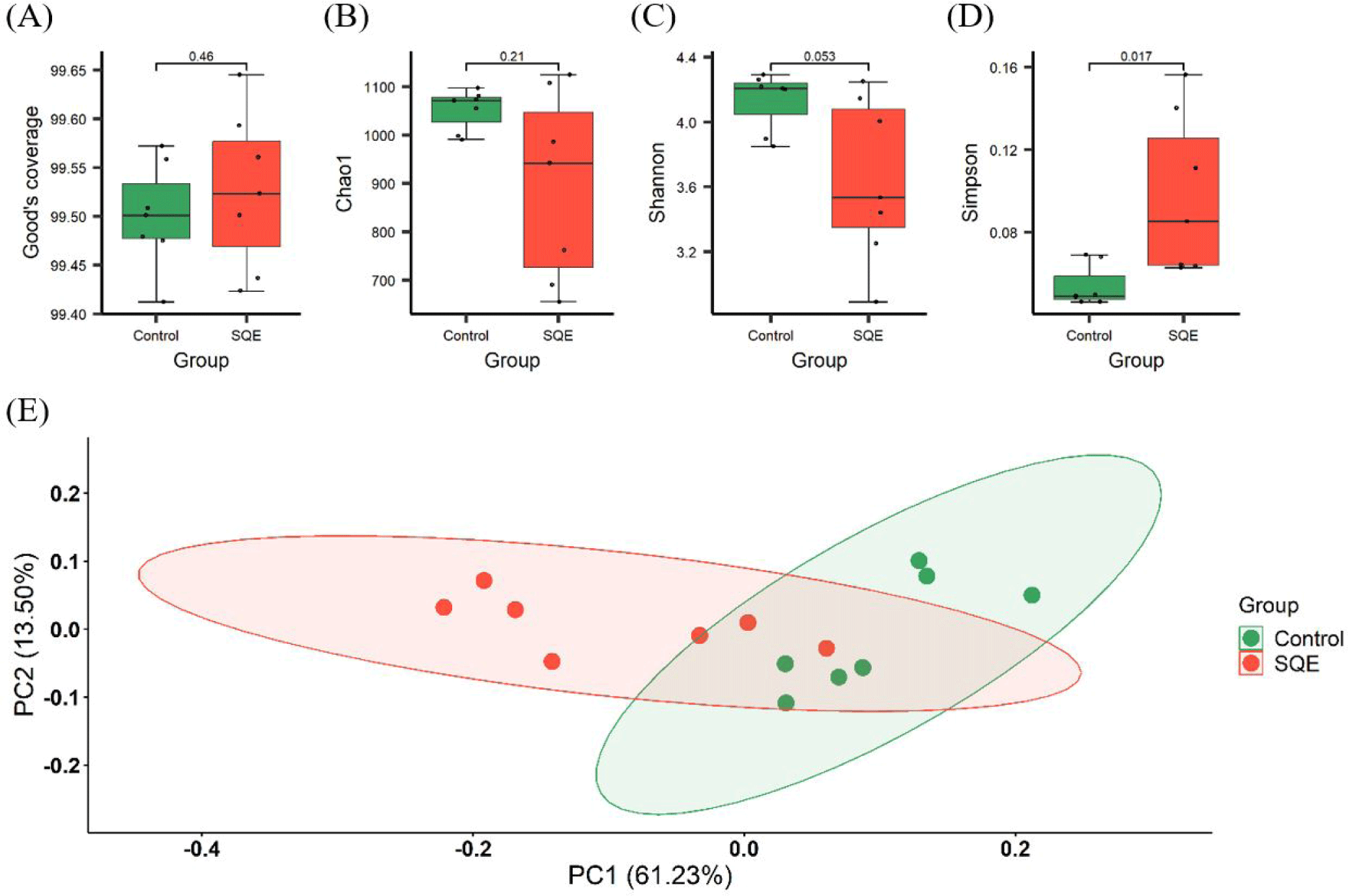
To assess the distribution of taxa between groups, we conducted alpha diversity analysis, which estimated the microbial richness and diversity. Good’s coverage index exceeded 99.4%, suggesting that the depth of 16S rRNA sequencing was sufficient to capture the fecal microbiota (Fig. 3A). The Chao1 index, a measure of species richness, showed similar observation values between groups (p > 0.05) (Fig. 3B). These findings indicate that SQE supplementation had a minimal impact on the count of different species present in the microbial communities. The diversity of species was evaluated using the Simpson and Shannon indices. The Shannon index decreased in the SQE group; however, the difference between observed values was marginal (p = 0.053) (Fig. 3C). In contrast, the Simpson index increased notably in the SQE group (p < 0.05) (Fig. 3D). Considering the diversity results, SQE supplementation affects the evenness of different species, leading to a less balanced composition of the microbiota. The PCoA plot of beta diversity evaluated by UniFrac distances showed dissimilarity in the microbial communities between the control and SQE groups (Fig. 3E).