
INTRODUCTION
Korean native cattle can be separated into four distinct breeds: Hanwoo (brown), Chikso (brindle), Heugu (black), and Jeju heugu (Jeju Black) [1,2]. Hanwoo constitutes a significant portion of the Korean native cattle population and is predominantly found on the Korean Peninsula, while Jeju Black cattle have been exclusively maintained on Jeju Island [3]. The Jeju Black cattle breed is believed to have been imported from the Mongol area in the 13th century and is currently classified as an endangered species due to a significant reduction in its population (Domestic Animal Diversity Information System, DAD-IS, FAO). In recognition of its cultural and ecological importance, Jeju Black cattle have been registered as a natural monument in Korea, and a specific breeding program has been implemented to conserve its population (Jeju Black Cattle, Cultural Heritage Administration, Korea, http://english.cha.go.kr/). Jeju Black beef is renowned for its high content of oleic acid, linoleic acid, and unsaturated fatty acids, making it a premium beef product in Korea [4]. However, its limited growth rate compared to that of Hanwoo and its habitat-specific characteristics have posed challenges for successful breeding programs [5]. While several studies have investigated the genetic differences between Jeju Black cattle and Hanwoo [2-4], research comparing their ruminal microbiome has been limited.
In Korea, beef feeding systems are structured into three distinct stages: the growing period, early fattening period, and late fattening period, each aimed at producing high-marbled beef. The growing period, which lasts up to 12 months of age, focuses on the development of bones, internal organs, and digestive organs, and is followed by the early fattening period, during which intramuscular fat deposition is induced through high concentrate feeding. The final stage, late fattening, concludes at 29–30 months, aiming to maximize intramuscular fat deposition by reducing the forage to concentrate ratio in the diet [6]. Increased concentrate feeding typically leads to a higher fermentation rate of ruminal microbiota, resulting in a rapid decrease in ruminal pH [7-9], elevated gas production, and propionate production [10-12].
In vitro digestibility can be influenced by various factors, such as the species of the rumen fluid donor animals and the type of feed used for their maintenance [13]. Differences in eating behavior, rumination, digestive tract physiology, ruminal retention time, and ruminal microbiome among donor animal species can impact the initial microbial and nutritional conditions of rumen inocula, ultimately influencing the overall fermentation characteristics in in vitro studies [13,14]. In vitro studies using rumen contents from different ruminant species as donors have demonstrated differences in nutrient degradability, microbial protein synthesis [15], and methane production [16,17]. Rumen samples from animals fed similar diets tend to exhibit comparable microbial compositions. For instance, consistent differences in gas production were observed in rumen samples collected from six different sheep fed the same diet, while in vitro degradation results were similar among nine different sheep receiving the same diet in the same environment [18,19].
In this study, we analyzed the differences in the ruminal microbiome and its relationship with ruminal fermentation under in vitro conditions using rumen inocula collected from both Hanwoo and Jeju Black cattle. Simultaneously, we investigated the effects of three different diets commonly fed during the growing, early fattening, and late fattening periods of beef cattle in Korea on the rumen fermentation characteristics, microbiota, and its functions.
MATERIALS AND METHODS
This study used six growth stages of Hanwoo and Jeju Black steers (body weight [BW], 400 ± 24.0 kg) to obtain rumen fluid samples. The steers were divided into two groups according to breeds of brown (n = 3; aged 16 months) and black (n = 3; aged 20 months) and housed in a tie stall at the same barn at the Seogwipo Korea Federation of Livestock Cooperatives Farm. This barn was equipped with an individual tie stanchion, feed bin, automatic waterer, and mineral block, and was bedded with wood shavings. Hanwoo and Jeju Black steers (one of each) were randomly assigned to three treatment groups (growing period total mixed ration [TMR], early fattening period TMR, and late fattening period TMR). The diet was formulated to meet the nutritional requirements of the National Research Council (NRC) [20] for beef cattle. The animals were adapted to the treatment diets for 2 weeks before the trial. The diet was fed twice daily during the adaptation period (at 09:00 and 17:00 h). Rumen fluid (approximately 200 mL) was collected from each Hanwoo and Jeju Black steer via a stainless steel stomach tubing at 2 h post-feeding and transferred to the laboratory using preheated thermos bottles. Each of the six rumen fluids was used for DNA extraction. Additionally, the rumen fluids of Hanwoo and Jeju Black cattle fed the early fattening period TMR were separately strained through four layers of cheesecloth and were maintained in a 39°C incubator before mixing with McDougall’s buffer solution [21]. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Chonnam National University, Korea (CNU IACUC-YB-2020-7).
Growing period, early fattening period, and late fattening period TMR diets were obtained from Seogwipo Korea Federation of Livestock Cooperatives Farm. All feed samples were dried and milled to pass through a 1-mm mesh screen in a Wiley mill (Model 4, Thomas Scientific, Swedesboro, NJ, USA). The dry matter (DM) was determined by drying the samples to a constant weight at 65°C. Samples were analyzed for crude protein (CP), crude fat (CF), and crude ash according to the methods described by the Association of Official Analytical Collaboration (AOAC) [22]. The moisture was calculated as × 100 by the AOAC [22]. Neutral detergent fiber (NDF) and acid detergent fiber (ADF) were evaluated as described by Van Soest et al. [23,24] using a fiber analyzer (ANKOM2000, ANKOM Technology, Macedon, NY, USA). The chemical compositions of the formulated TMR diets are shown in Table 1.
McDougall’s buffer solution was heated at 39°C, flushed with O2-free CO2 for 20 min, and then mixed with the rumen fluid in a 2 : 1 (vol : vol) ratio under anaerobic conditions. Next, 40 mL of the buffered rumen fluid was dispensed into a 120 mL gas-tight serum bottle filled with bubbled O2-free CO2 and was then flushed into the headspace of the serum bottles, which were then closed with a butyl rubber stopper containing a rubber septa with an aluminum seal, respectively, and incubated at 39°C. The DAISYII apparatus contained four 4-L digestion vessels, which slowly rotated in a digestion chamber that was maintained at 39.5°C. Samples to be analyzed were heat sealed into ash-free and N-free filter bags and inserted into the digestion vessels. The in vitro experiment was conducted for 3 h and 24 h under a 2 × 3 factorial arrangement using two breeds (Hanwoo and Jeju Black) and three substrates (growing period TMR, early fattening period TMR, and late fattening period TMR) as factors.
In vitro total gas measurements were calculated from the headspace gas pressure measured by a pressure transducer (Sun Bee Instrument, Seoul, Korea). The pH of the rumen fluid was measured with a pH meter (Orion Star A211 bench-top pH meter, Thermo Scientific, Bremen, Germany) and the residual rumen fluid samples were stored at –20°C immediately for DM digestibility and volatile fatty acids (VFA) and ammoniacal-N (NH3-N) analysis. After thawing, 10 mL of the sample was mixed with 1 mL of HgCl2 2% (wt/vol) solution and briefly centrifuged at 2,000×g for 10 min at 4°C to remove feed particles. The supernatants were then used for DNA extraction and VFA and NH3-N analysis. The VFA concentration was determined using gas chromatography (Varian CP-3800, Varian, Walnut Creek, CA, USA). The NH3-N concentration was determined using a Multiplate spectrophotometer (Benchmark PlusTM, Bio-Rad, Tokyo, Japan) at 625 nm as described by Chaney and Marbach [25].
Total community DNA was extracted from 42 rumen fluid samples (six from donor animals and 36 from the in vitro rumen fermentation experiment) using a Mini-Beadbeater-16 (BioSpec Products, Bartlesville, OK, USA) and the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) [26]. To amplify 16S rRNA gene amplicons, a library was generated from each DNA sample using the 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTA CHVGGGTATCTAATCC-3’) primers targeting the V3-V4 region, and was sequenced on the MiSeq platform (Illumina, San Diego, CA, USA). Pairs of reads were merged using the Fast Length Adjustment of SHort reads (FLASH) program [8].
The sequences were analyzed using QIIME2 (version 2022.11) [27]. Primers were trimmed using Cutadapt [28]. After trimming off the adapter and primer sequences, DADA2 was performed for filtering by quality score (≥ 25) and removal of chimeric sequences [29].
Bacteria and archaea were classified using scikit-learn [30] with the weighted SILVA reference database (v.138) [31]. Further taxonomic filtration was done to remove the amplicon sequence variants (ASVs) labeled as ‘unassigned,’ ‘chloroplast,’ or ‘mitochondria.’ The average rarefied abundance table was made by averaging the 1,000 times repeated rarefaction outputs at 1,170 ASVs computed by q2-repeat-rarefy [32].
α-diversity measurements including species richness (observed ASVs and Chao1 estimates), evenness, Shannon’s index, and Simpson’s index were calculated based on the aforementioned repeatedly rarefied ASV abundance table. Comparison of the overall microbiotas between the Hanwoo inoculum-treated group (HWG) and Jeju Black inoculum-treated group (JBG), and between the growing, early-fattening, and late-fattening diets were done using principal coordinates analysis (PCoA) based on the Bray-Curtis distance matrix [33].
Functional microbial features were predicted from the 16S ASVs using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2) [34]. The normalized counts of the predicted enzyme commission (EC) numbers were used to define the overall functional dissimilarities between HWG and JBG, and between diets. Principal components analysis (PCA) based on the Bray-Curtis distance matrix was used to analyze overall functional dissimilarities. PCA outputs were visualized using the ggfortify package of R (4.2.2) [35].
Fermentation data and α-diversity metrics results were statistically analyzed separately for 3 h and 24 h of the in vitro incubation using the GLIMMIX procedure of SAS 9.4 (SAS Institute, Cary, NC, USA), where inocula (HWG and JBG), diets, interaction between inoculum, and diet effects were set as fixed effects. Permutational multivariate analysis of variance (PERMANOVA) was used to statistically analyze the overall microbiotas and their functional dissimilarities between time, inoculum, and diet effects with interactions using vegan and the pairwiseAdonis package of R (4.2.2) with 9,999 random permutations followed by multiple-test corrections with the Benjamini-Hochberg correction [36]. For statistical analysis of microbiome data, major microbial features including classified taxa at the phylum, family, and genus level and microbial functions represented by predicted ECs selected when the average relative abundance of each feature was over 0.1% across all samples were mainly discussed in this study. Differentially abundant microbial phyla, families, genera, and EC numbers were determined using Microbiome Multivariable Associations with Linear Models 2 (MaAsLin2) [37] with the Benjamini-Hochberg corrected p-value (q-value) 0.05 as the cutoff for significant differences. Relative abundances of microbial taxa and predicted EC numbers were normalized with a centered-log ratio and used for the MaAsLin2 analysis. Except for the relative abundance data, statistical significance was declared at p ≤ 0.05.
RESULTS
In vitro fermentation characteristics according to inoculum and diet effects are shown in Table 2. The pH was high in the JBG at both incubation times (p < 0.0500). NH3-N was higher in the JBG after 3 h of incubation (p = 0.0147), while the opposite was found after 24 h (p = 0.0273). Total gas production and dry matter digestibility (DMD) was higher in HWG at both incubation times (p < 0.0500) but the total VFA concentration was enriched more in the HWG at only the earlier incubation period (p < 0.0001). Among the major VFAs, the molar proportion of acetate and valerate showed opposite trends at 3 h of incubation, while no significant differences were found in any VFA profile at 24 h.
At 3 h of incubation, the total gas production was higher in the growing diet compared to that of the late fattening diet. Additionally, the DMD were significantly lower in the early fattening diet with a significant interaction between the inoculum and diet (p = 0.0042), while the total VFA concentration was only high in the late fattening diet. For each VFA profile, no significant diet effect was found except with valerate, and a significant difference was seen between the early and late fattening diets. At 24 h of incubation, the pH, gas production, DMD, and total VFA concentration did not differ by diet, and only NH3-N was significantly higher in the early fattening diet compared to that of the other diets. Additionally, opposite profiling was found between propionate and butyrate. Increasing the molar proportion of propionate at the end of the fattening period resulted in the lowest A:P ratio by the late fattening diet. For all measurements at 24 h of incubation, no significant interaction effect between the inoculum and diet was found (p > 0.100).
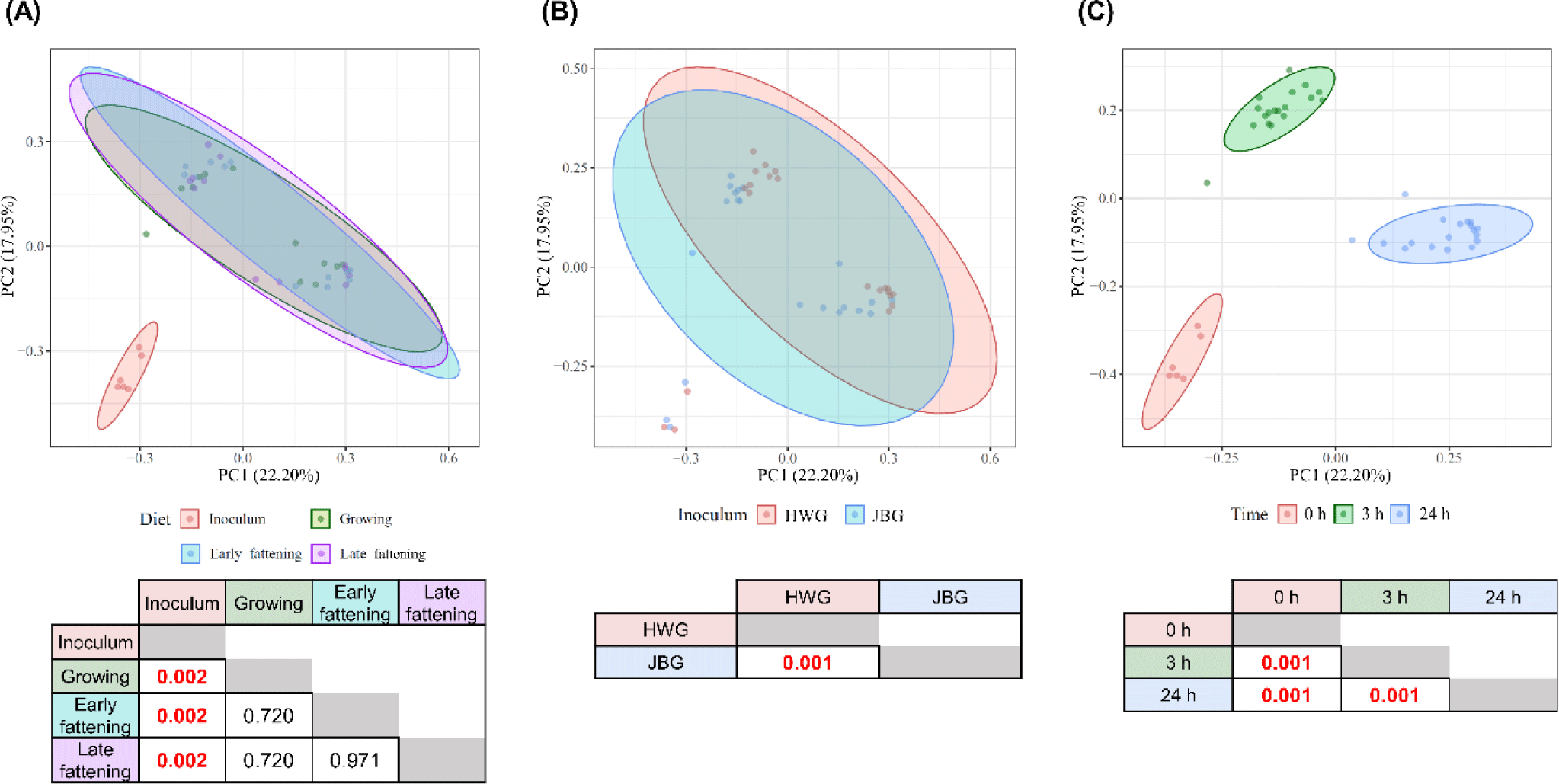
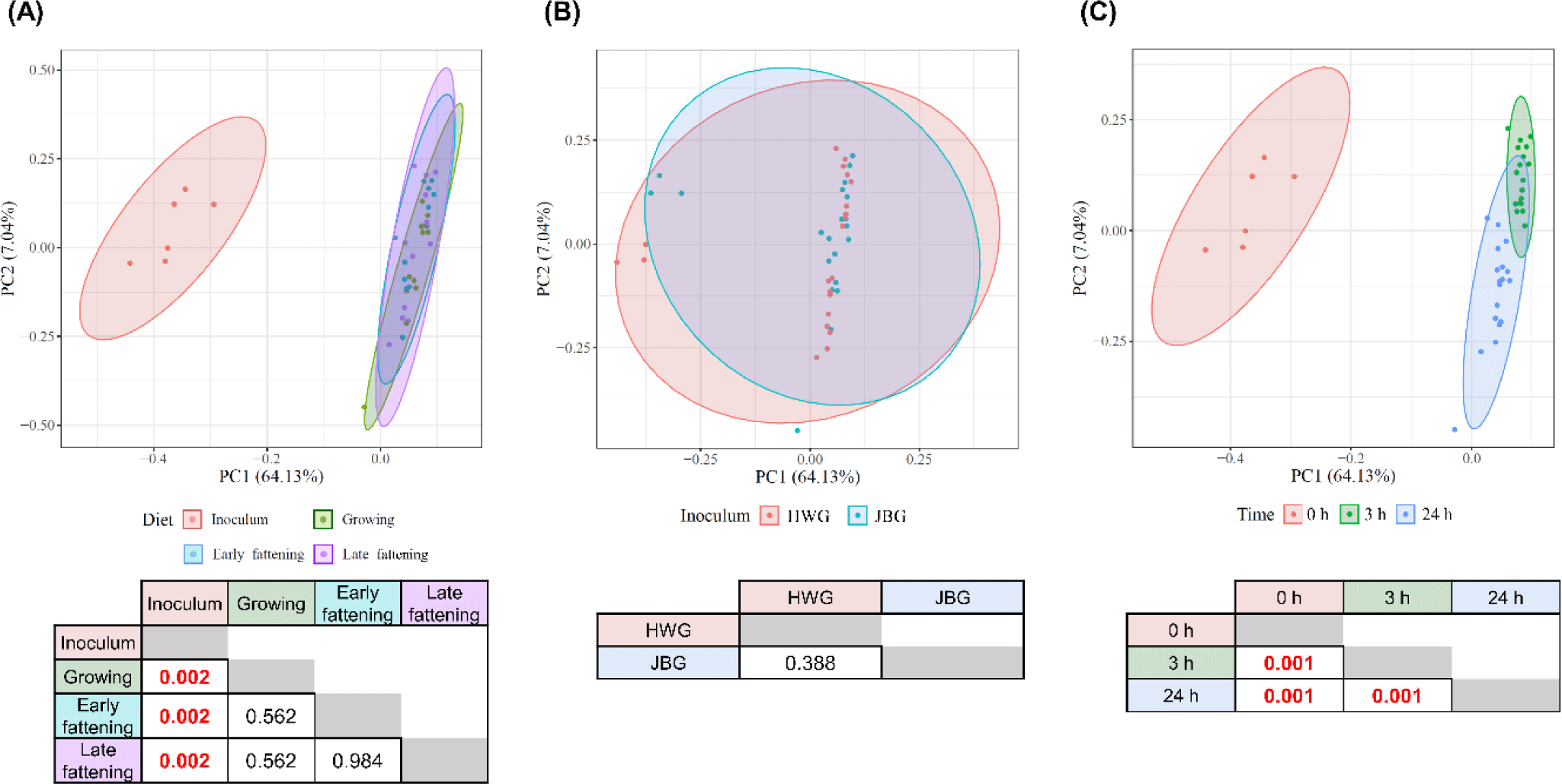
No significant differences were found in any of the analyzed a-diversity measurements between both inoculums (p > 0.1) (Table 3). At 3 h of incubation, the observed features of the HWG were higher than those of the JBG (p = 0.0268), but after 24 h of incubation, the difference by inoculum treatment was offset. No significant diet effects were found in any α-diversity measurements. Overall, the microbial community structures significantly differed by incubation time and inoculum (q < 0.01) using the Bray-Curtis distance matrix with no significant diet and interaction effects seen among the fixed effects (p > 0.05) (Fig. 1).
Among the major taxa across all 42 samples, at the phylum level, Firmicutes (80.44%) was primarily dominant, followed by Proteobacteria (8.48%) and Bacteroidetes (2.55%). Lachnospiraceae (17.54%), Ruminococcaceae (14.31%), and Planococcaceae (10.56%) were the three most dominant families, while Solibacillus (9.77%), Succinivibrio (7.40%), and an unclassified genus within Lachnospiraceae (6.99%) were found as major genera across all samples (Supplementary Data Tables S1 and S2).
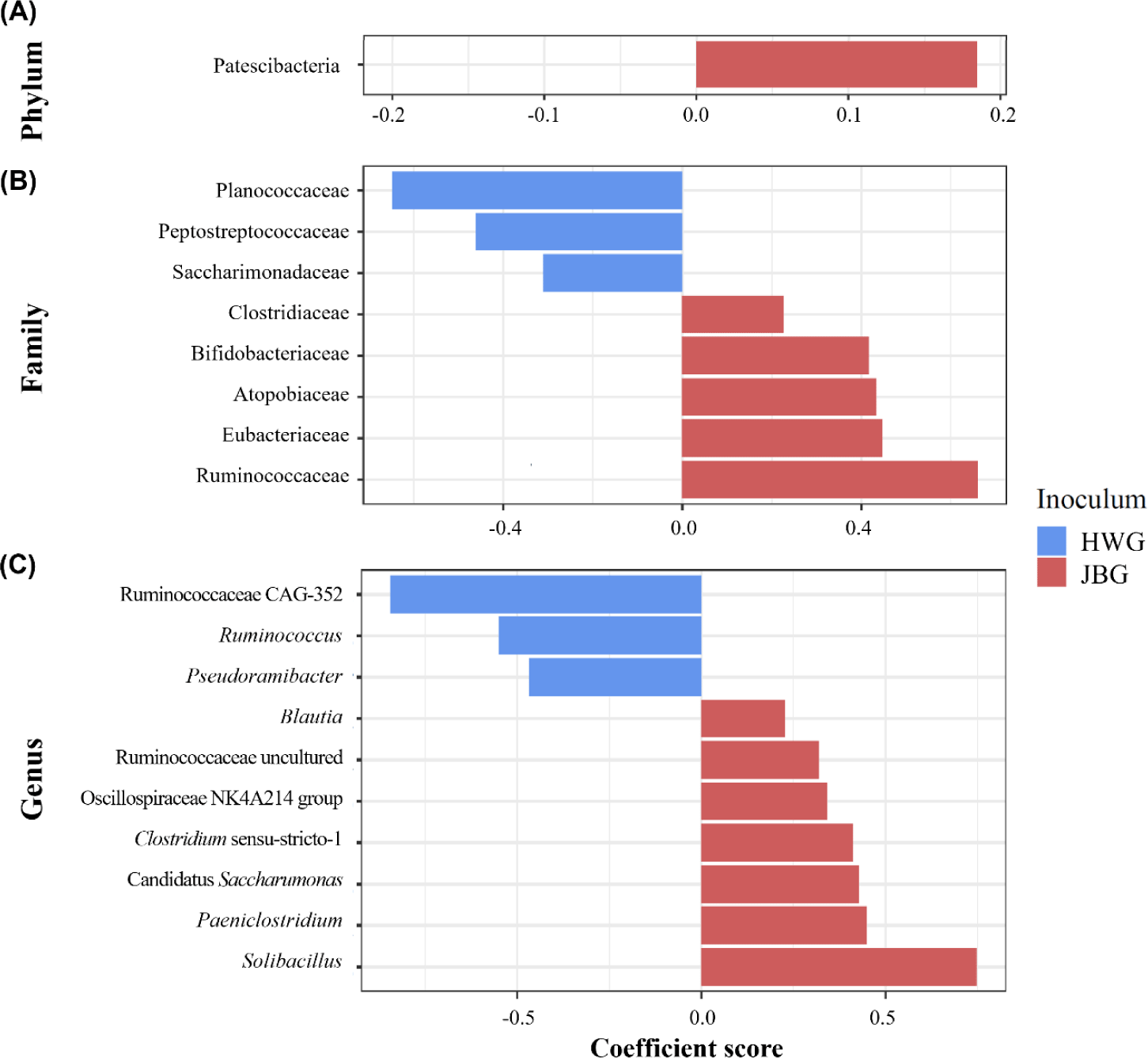
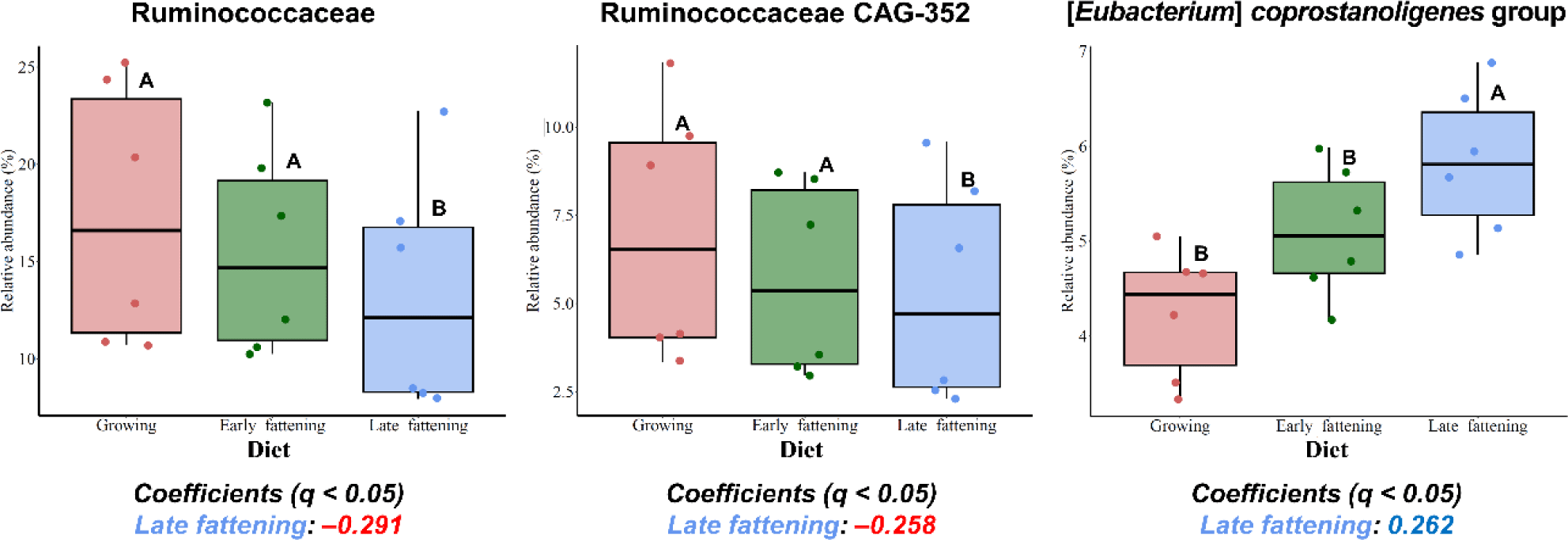
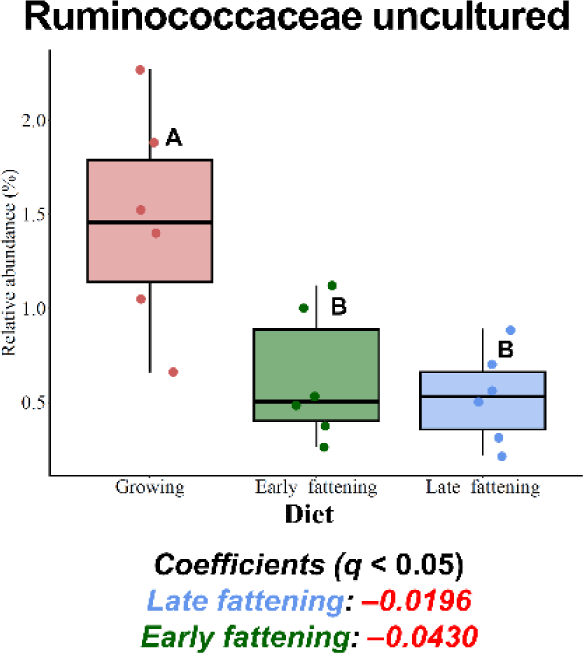
Among the major phyla selected in this study, only Patescibacteria was more enriched in the JBG than in the HWG (6.39% vs. 3.86%, respectively) after 3 h of incubation (Fig. 2A). Among the major families, Planococcaceae, Peptostreptococcaceae, and Saccharimonadaceae were differentially abundant in the HWG, while Clostridiaceae, Bifidobacteriaceae, Atopobiaceae, Eubacteriaceae, and Ruminococcaceae were differentially abundant in the JBG (Fig. 2B). At the genus level, Ruminococcus, Pseudoramibacter, and Ruminococcaceae CAG-352 were more enriched in the HWG, while Blautia, Clostridium sensu-stricto-1, Paeniclostridium, Solibacillus, Oscillospiraceae NK4A214 group, and an uncultured genus within Ruminococcaceae were more enriched in the JBG (Fig. 2C). At the family level, Ruminococcaceae was significantly enriched in the growing diet, and at the genus level, Ruminococcaceae CAG-352 was also enriched in the growing diet, while the [Eubacterium] coprostanoligenes group was enriched in the late fattening diet (Fig. 3).
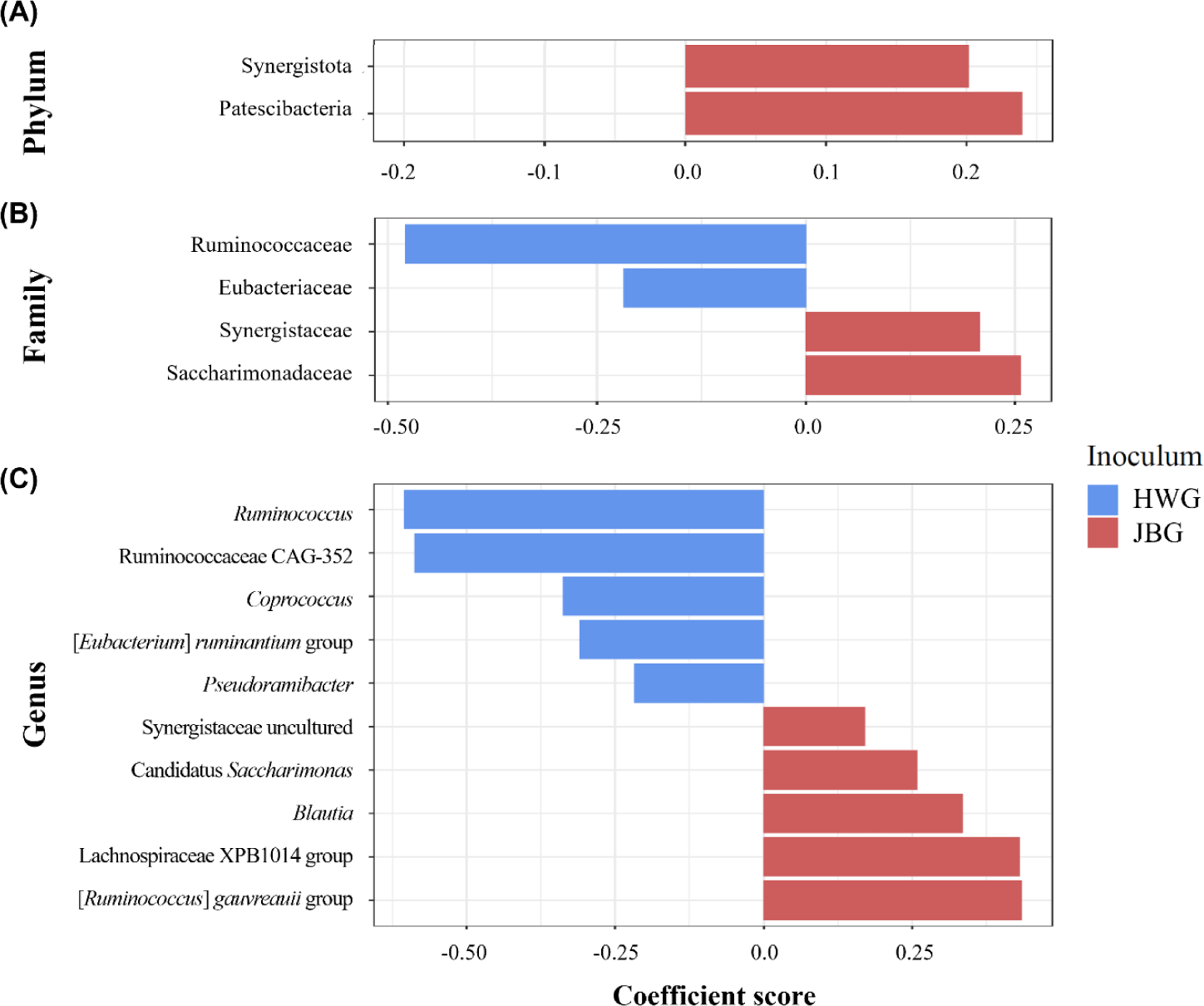
Among the major phyla, Patescibacteria and Synergistota were differentially abundant in JBG (Fig. 4A). At the family level, Ruminococcaceae and Eubacteriaceae were significantly enriched in the HWG, while Synergistaceae and Saccharimonadaceae were significantly enriched in the JBG (Fig. 4B). At the genus level, the predominance of Ruminococcus, Ruminococcaceae CAG-352, Coprococcus, [Eubacterium] ruminantium group, and Pseudoramibacter were confirmed in the HWG, while Candidatus Saccharimonas, Blautia, Lachnospiraceae XPB1014 group, [Ruminococcus] gauvreauii group, and uncultured genera within Synergistaceae were predominant in the JBG (Fig. 4C). At the genus level, the relative abundance of the uncultured genus within Ruminococcaceae was significantly higher in the growing diet compared to that of the fattening period diets (Fig. 5).
Based on the predicted microbial functions represented by the EC numbers analyzed using PCA visualization and PERMANOVA, it was shown that only the in vitro incubation time significantly affected the overall functional microbial community and all pairwise comparisons between the three time points differed significantly (q = 0.001), while the inoculum and diet effects did not show a significant difference (q > 0.05) (Fig. 6). No significant interaction effect was found between the time, inoculum, and diet effects (q > 0.05).
Comparative analysis of functional features to analyze inoculum and diet effects during in vitro incubation was done using MaAsLin2 based on the relative abundance of the predicted microbial EC numbers. Among the major EC numbers, 190 features differed by inoculum effect at 3 h of incubation (Supplementary Data Table S3). After 24 h of incubation (Table 4), seven specific EC numbers including four transferases (EC 2), two hydrolases (EC 3), and one lyase (EC 4) differed significantly by inoculum effect. The discrepancy pattern of those seven specific EC numbers was also similar at 3 h of incubation.
Five EC numbers differed by diets (Table 5). Among these, one oxidoreductase (EC 1) and two transferases (EC 2) were significantly enriched in the late fattening diet, while lyase (EC 4) and ligase (EC 6) were enriched in the growing diet. These results were obtained only at 3 h of incubation, and there were no significantly different functional features at 24 h of incubation.
DISCUSSION
The present study investigated the impact of different inocula on the ruminal microbiota In vitro. Our results demonstrated significant differences in the microbial community composition between two inoculum-treated groups, as evident from the distance matrix analyzing the compositional dissimilarity. These findings are consistent with previous studies, which highlighted how the overall microbial community structures in rumen fluid can be influenced by various factors, including breed and cattle subspecies [38,39]. In this experiment, the donor animals were provided with an early fattening period diet and raised on Jeju Island. Rumen fluid samples were collected through stomach sampling techniques. As noted by Hagey et al. [40], the microbial composition can exhibit variations depending on the sampling method employed. At the phylum level, it was observed that Firmicutes increased, and Bacteriodota (Bacteriodetes) decreased when compared to rumen fistula fluid filter through cheesecloth. In a separate study, certain members of the Firmicutes phylum, such as Christensenellaceae and Lachnospiraceae, were reported to have potentially higher abundance when sampling liquid-based fluid via stomach sampling [41]. Furthermore, numerous factors, including geographical location and diet, have been identified as influential contributors to the microbial diversity and quantity within the digestive tract [42]. Under high-concentrate feeding conditions, Firmicutes accounted for over 70% of the detected microbes. At the genus level, Prevotella experienced a significant decrease under high-concentrate feeding conditions when compared to low-concentrate diet [43].
During the early incubation time, there were only transient differences in richness, while more pronounced compositional differences emerged later, resulting in a significant disparity in the overall microbial community between the two inocula-treated groups. This effect can be attributed to the distinct physical and physiological traits of the highly domesticated Hanwoo cattle and the relatively native Jeju Black cattle. Gut size and ruminal passage rate are known to influence the rumen microbial composition [44,45], possibly leading to the acquisition of distinct rumen microbiota by the two groups.
At the family and genus levels, certain members of the ruminal microbiota were significantly enriched in the HWG. Notably, carbohydrate fermenting bacteria such as Ruminococcus, Pseudoramibacter, and Ruminococcaceae CAG-352 from the Firmicutes phylum were identified. Ruminococcus species are well-known cellulolytic bacteria, including R. flavefaciens and R. albus [46-48]. Pseudoramibacter consumes glucose or pyruvate and produces acetate [49], while uncultured genus CAG-352 is primarily fibrolytic [50]. These genera likely contributed to fibrolysis more rapidly in the HWG, resulting in a significantly higher total VFA concentration and acetate proportion during the early incubation time. The [Eubacterium] ruminantium group, which was mainly found in rumen fluid, is hemicellulolytic (mainly xylan) [51,52] and produces lactate, butyrate, and formate [53]. Coprococcus ferments aromatic compounds into organic acids such as acetate and benzoate [54]. C. catus [55], C. eutactus, and C. comes [56] are reported to produce succinate, which is a precursor of propionate. These two genera, which appeared to be enriched only at the later incubation time in the HWG, may have increased production of other VFAs rather than acetate, thus offsetting the difference in VFA profiles at 24 h of incubation.
Similarly, certain microbial taxa were enriched in the JBG, including Patescibacteria (also known as Candidate Phyla Radiation [CPR]), which possesses surface proteins facilitating its attachment to other microorganisms like bacteria and methanogens [57]. Additionally, Synergistota can oxidize acetate into H2 and CO2 [58] and may provide short-chain fatty acids and sulfate to methanogens or sulfate-reducing bacteria [59]. Given the differences in the grazing environment and degree of domestication for Jeju Black cattle, methane production might be higher compared to that of Hanwoo [60]. However, methane production was not measured in this study, and further research is required to directly correlate the abundances of these enriched microbial taxa with methane production.
The analysis of relative abundance in 3 h and 24 h samples did not reveal significant differences in the in vitro fermentation characteristics between the two groups. However, it was observed that certain bacterial taxa associated with the JBG showed fermentative traits that might explain the distinct VFA profiles observed, especially during the early stages of incubation. Notably, the Blautia species including B. hydrogenotrophica, B. schinkii, and B. producta were found to produce acetate, lactate, or succinate [61]. Similarly, the [Ruminococcus] gauvreauii group was identified as a major acetate producer [62,63]. Candidatus Saccharimonas was positively correlated with propionate production in dairy cows [64], while the Oscillospiraceae NK4A214 group was negatively correlated with acetate and propionate proportions in young goats [65]. Additionally, the abundance of Solibacillus was found to be positively correlated with valerate under acidosis conditions in dairy cows [65].
Taken together, by using different inocula, the differentially abundant bacterial genera have some features that can make differences in in vitro fermentation and that might be responsible for the VFA profiles at the beginning of the incubation period and the overall in vitro digestibility.
The present study investigated the impact of different diets on the ruminal microbiota. Surprisingly, no significant differences were observed in the overall ruminal microbiota in response to the three different diets. This lack of variation was reflected in all analyzed α-diversity measurements, indicating that the number of microbes, evenness, and overall diversity of the ruminal microbiota remained consistent under in vitro conditions. While the diets were formulated to meet the nutritional requirements for each stage of growth and fattening, the relatively small variation in the chemical composition of the experimental diets may have resulted in only mild differences during in vitro incubation. This assumption is further supported by the lack of differences in digestibility measurements and minor variations in fermentation characteristics. It is important to consider these subtle differences when interpreting the microbiome data in this study.
Although limited studies have explored the relationship between the [Eubacterium] coprostanoligenes group and the forage-to-concentrate ratio, a previous study involving feed additives reported a positive correlation between this genus and ruminal pH [66]. The prevalence of the [Eubacterium] coprostanoligenes group in the late fattening diet could have influenced the ruminal pH, thereby potentially explaining the lack of significant differences in ruminal fermentation characteristics during later incubation times. The role of this bacterial group in pH regulation highlights the complexity of ruminal microbial interactions and their potential impact on the fermentation process.
Ruminococcaceae is a prominent taxon within the Firmicutes phylum in the rumen [67,68] known for its cellulolytic and fibrolytic capabilities [69,70]. In the present study, Ruminococcaceae exhibited differential abundance in the growing diet, which contained more fiber-rich ingredients such as timothy hay and alfalfa. Interestingly, uncultured and tentative genera within the Ruminococcaceae family, namely Ruminococcaceae CAG-352 and an uncultured Ruminococcaceae genus, were enriched at 3 h and 24 h of incubations, respectively. Previous research has shown that some uncultured/unclassified genera within Ruminococcaceae, including Ruminococcaceae UCG-011 and UCG-010, were enriched in the intensive forage feeding condition [70,71]. The high relative abundance of Ruminococcaceae CAG-352 at 3 h of incubation, which ranged from 29.0% to 48.6%, may exert a notable impact at the family level. The presence and activity of these specific taxa could be linked to fiber digestion.
In contrast to the overall microbial community, the functional microbial community did not differ significantly due to the inoculum effect. These findings are consistent with previous studies in the rumen, highlighting that different microbial communities may share similar functional potential [72].
Notably, six EC numbers were enriched in the HWG, including 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (EC:2.7.7.60), phosphoribosyl-adenosine triphosphate (ATP) diphosphatase (EC:3.6.1.31), and tryptophan synthase (EC:4.2.1.20), which contain nucleic acid, phosphate, or nucleoside triphosphate (NTP), and were involved in the biosynthesis of secondary metabolites, and beta-ketoacyl-[acyl-carrier-protein] synthase II (EC:2.3.1.179), which was involved in fatty acid biosynthesis. However, the predicted microbial function difference was not related to fermentation characteristics or the ruminal microbiota diversity. Additionally, the enrichment of alpha-amylase in the HWG did not lead to significant changes in glucose fermentation or ruminal pH, suggesting that other factors may play a more significant role in determining these outcomes.
S-ribosylhomocysteine lyase (EC:4.4.1.21) and asparagine synthase (glutamine-hydrolyzing) (EC:6.3.5.4) were found to be enriched in the growing diet at 3 h of incubation. These enzymes are involved in protein biosynthesis, specifically the synthesis of cysteine, methionine, aspartate, and glutamate. The enrichment of these microbial enzymes in the growing diet at an earlier incubation time related to protein biosynthesis resulted in the numerically and significantly enhanced nitrogen utilization efficiency at both 3 h and 24 h of incubation, respectively. Previous studies have reported a negative relationship between NH3-N concentration in the rumen and the number of microbial proteins derived from non-protein nitrogen [73,74]. Microbial proteins synthesized from multiple nitrogen sources serve as a crucial source of amino acids for ruminants [75,76]. The increased nitrogen utilization efficiency observed in this study suggests that the diet for the growing period potentially promoted beef cattle growth by delivering a greater amount of nitrogen sources to the small intestine [77].
In conclusion, this study used Jeju Black and Hanwoo cattle, both of which belong to the Bos primigenius species, as rumen fluid donors for an in vitro fermentation experiment with three different diets corresponding to the fattening stages of beef cattle. Our results suggest that ruminal microbiota differences present in the two different inocula may influence the early stages of fermentation and microbial composition. However, these differences were only partially offset after 24 hours of incubation. While the inoculum significantly affected the overall microbial community, it did not seem to exert a substantial impact on the functional microbial community. Among the differentially abundant taxa affected by the inoculum effect, Patescibacteria, Ruminococcus, Pseudoramibacter, Blautia, and Solibacillus could potentially contribute to the observed differences in rumen fermentation characteristics and overall microbial community structure. Dietary effects were not significant based on the alpha- and beta-diversity measurements, which showed minor differences in the bacterial taxa. However, it is essential to consider that the rumen fluid used in the experiment came from donor animals that were adapted to the diets for only 2 weeks. Thus, the original differences present in the inoculum, as indicated by specific microbial taxa, overall diversity, and functional features, should be carefully considered while interpreting the potential diet effects. In conclusion, this study provides valuable insights into the rumen microbiome and functional characteristics of Hanwoo and Jeju Black cattle.
SUPPLEMENTARY MATERIALS
Supplementary materials are only available online from: https://doi.org/10.5187/jast.2023.e109.