
INTRODUCTION
Gut microbiota is generally known to play a significant role in maintaining host health and metabolism [1]. It is also important for maintaining growth performance of animals. The pig gut is inhabited by a large and varied population of bacteria, archaea, viruses and eukaryotes like fungi. It is estimated that a mammalian digestive tract contains approximately 1014 bacteria [2,3]. The gut microbes of pigs live in close contact with each other and share a set of mutual and symbiotic relationships. It has been hypothesized that microbiome benefits to animal health and growth performance by limiting potential pathogens to colonize the gastrointestinal tract and thus preventing pathogen infections [4]. It has also been shown that experimental oral inoculation with specific pathogens lead to change in the pig’s gastrointestinal microbiome [5]. A deeper comprehension of the functions played by the microbiome is likely to help us define a healthy microbiome, understand disease pathophysiology, and maybe develop new disease-control tactics and growth enhancement strategies [6]. In addition, pork is one of the most consumed meats in the world, and hence research on pig’s intestinal microbes and host metabolism will greatly promote capacity of pig production.
Therefore, a better understanding of these aspects could provide information on healthy and efficient pig production, as well as advance our knowledge regarding the relation between the gut microbiome and microbiome-host crosstalk mechanisms. More importantly, knowledge of the microbiota, host health and metabolism can facilitate the development of precise growth factors to boost up pig growth. Thus, the present study investigated microbiome changes from 70 days to 160 days of age based on the varied age of shipment in the three farms. The microbiome changes from growing stage to finishing stage with respect to the difference in shipment age were also followed.
MATERIALS AND METHODS
We used crossbred pigs (Duroc × [Landrace × Yorkshire]) that were bred in 3 different farms; two in Gyeonggi-do and one in Chungcheongnam-do, South Korea. The three selected farms were similar in pig breed, nutrition (feed and feed additives), and size, but differed in farm facilities, hygiene practices, and management. Based on these criteria we ranked them as follows. 1) excellent facilities and management (farm D; D), 2) average (farm T; T) 3) below average (farm J; J). When pigs reached average market weight of 118 kg, the average age of pigs in three different farms were < 180 days (D), about 190 days (T), and > 200 days (J), respectively. The pigs were fed a conventional wheat-soybean meal basal diet that complied with the National Research Council (NRC) standards. A total of 36 fresh fecal samples were collected, feces from rectum of 3 pigs from each farm of age of 70, 100, 130 and 160 days.
Total DNA extraction was performed using the QIAamp Fast DNA Stool Mini Kit (QIAGEN, Hilden, Germany) according to the instructions provided by the manufacturer using 200 mg of feces per sample [7]. During the total DNA extraction step, one major modification was an addition of a steel bead beating step in the beginning of DNA extraction. The primer set used for amplifying the hypervariable regions V5-V6 of the bacterial 16S rRNA gene was fwd: 799F-mod2 (5’ AACMGGATTAGATACCCKGGT 3’) and rev: (5’ GCAACGAGCGCAACCC 3’), resulting a PCR product of around 315 bp [8]. The size of the amplicons was validated by gel electrophoresis. DNA purification was done with Wizard SV Gel and PCR clean up purification system (Promega, cat. No. A1331) following the manufacturer’s guidelines. Then, the purified DNA was stored at −20°C until further usage.
Amplicon libraries were prepared by 5’ and 3’ adapter ligation following random fragmentation of DNA samples. In this step, the Nextera XT index kit was used to connect the dual index and Illumina sequencing adapter. PCR conditions applied were, initial denaturation (3 minutes at 95°C), 8 amplification cycles (95°C 30 seconds, 55°C 30 seconds, 72°C 30 seconds), and final extension (72°C 5 minutes). We quantified and pooled the final products using PicoGreen, and confirmed the library size using TapeStation DNA Screen Tape D1000 (Agilent, Santa Clara, CA, USA). Amplicons were sequenced using Illumina Miseq reagent kit v3. 2 × 300 bp paired-end sequencing (BRD Korea, Hwaseong, Korea).
To evaluate the pig fecal microbial diversity and community structure, we used 16S rRNA gene sequence analysis. The 16S rRNA gene sequences were analyzed using the Mothur software package (Version 1.40.5) following the analysis protocol of Miseq SOP (http://www.mothur.org/wiki/Miseq_SOP) with some modifications. 16S rRNA gene sequences were trimmed with following parameters (qaverage = 27, maxambig = 0, maxhomop = 8, minlength = 100, maxlength = 700). De novo operational taxonomic unit (OTU) clustering with an OTU definition at an identity cutoff of 97% was conducted using QIIME (Quantitative Insights into Microbial Ecology) software package (version 1.9.1) [9]. Using Analysis of Variance (ANOVA) in Statistical Analysis of Metagenomic Profiles (STAMP) software v2.1.3 and R package MicrobiomeAnalystR, the observed OTU, Chao1, Shannon, and Simpson indices were calculated. The nonparametric Kruskal-Wallis test was used to calculate significant differences in alpha diversity between groups. The significant difference threshold was set to p < 0.05. Principal Coordinates Analysis (PCoA) plots were generated at the OTU level based on weighted and unweighted UniFrac distance metrics. Beta diversity was measured using analysis of similarity (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA) based on weighted and unweighted UniFrac distance metrics.
RESULTS
A total of 11,993,437 reads ranging from 111,602 to 255,215 reads per sample were generated after the sequencing of 16S rRNA genes.
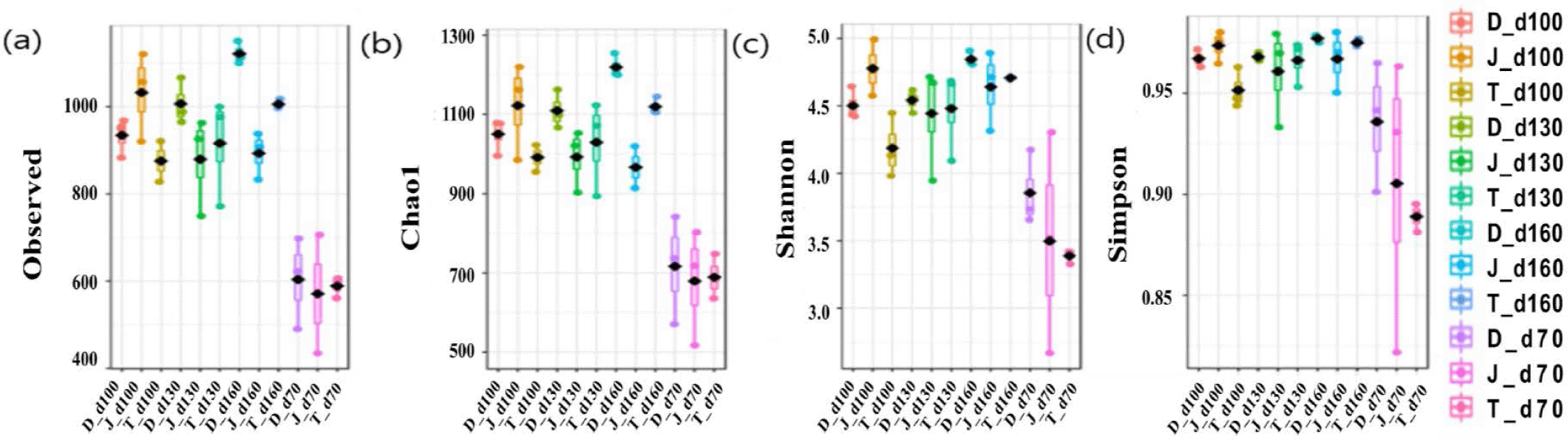
The microbial diversity in fecal samples was measured using the Chao1 (species abundance estimator), observed number of OTUs, and Shannon and Simpson indices (considering species uniformity). Overall, the alpha diversity indices showed that the gut bacterial diversity increased over time as pigs aged regardless of the farms (Fig. 1).
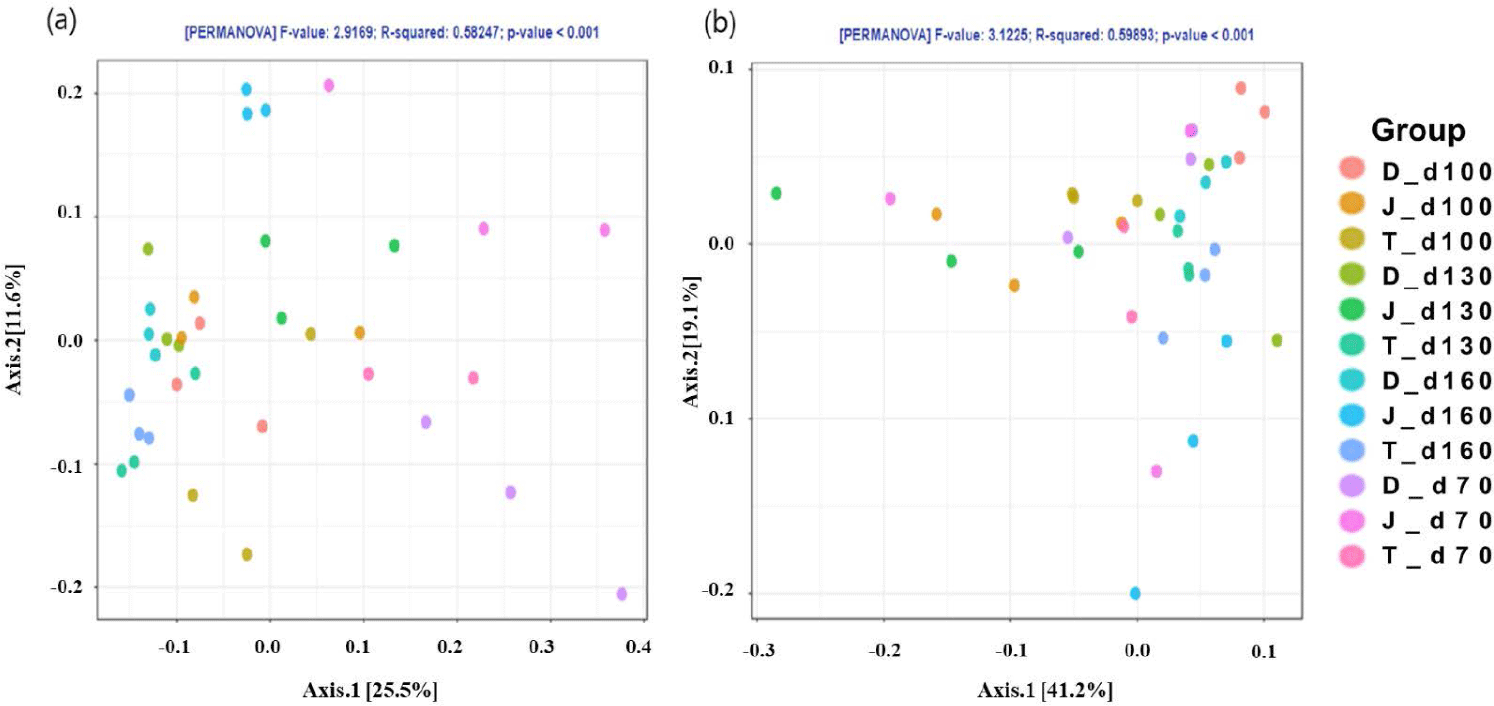
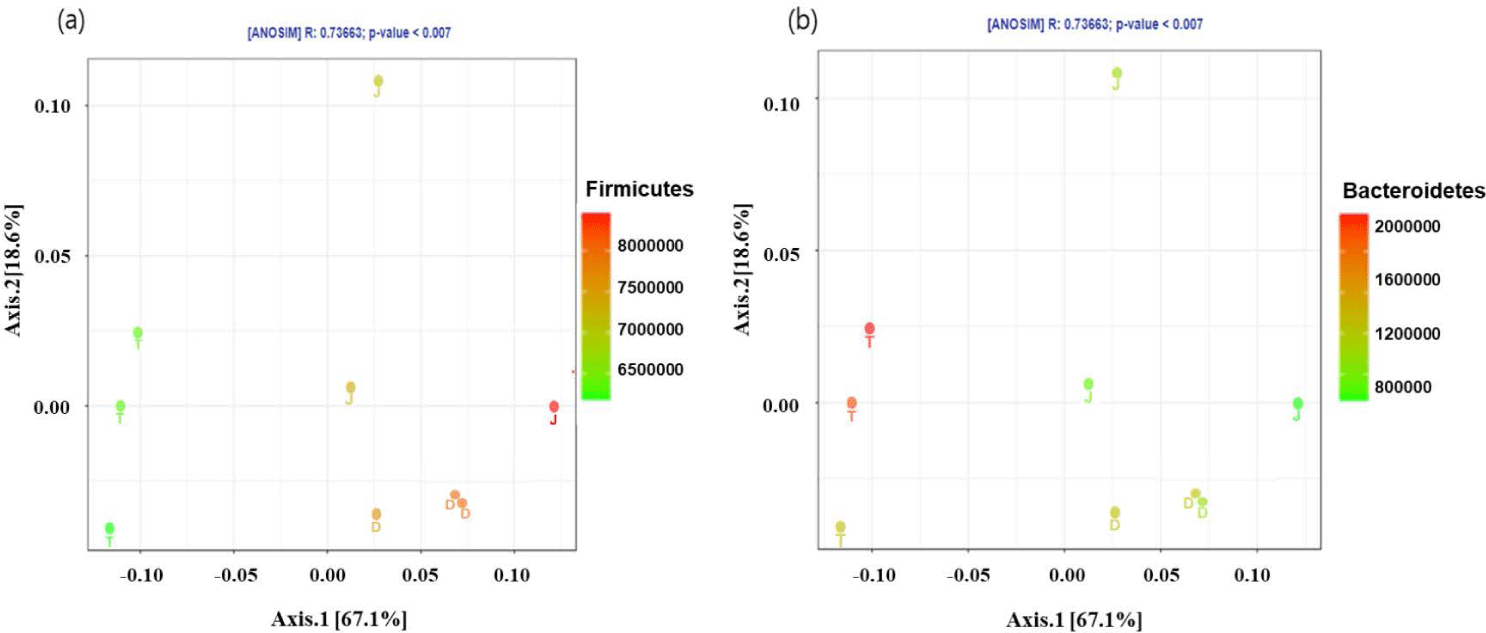
The PCoA plot did not show significant isolation of the microbial community among the groups, which was confirmed by PERMANOVA using both the weighted (Fig. 2A) and unweighted (Fig. 2B) UniFrac metrics (p > 0.05) (Fig. 2). However, the PCoA plots based on the weighted UniFrac and unweighted distance metrics using only the intestinal microbiota of Firmicutes and Bacteroidetes at the 160-day-old showed distinct clustering (Fig. 3).
The relative abundance of different bacterial taxa at the phylum level among the three groups at 70, 100, 130, and 160 days of age was shown in Fig. 4A. Regardless of age, the microbial communities were composed predominantly of phyla Firmicutes and Bacteroidetes. At 70 days of age, the most prevalent bacteria were Firmicutes, and the relative abundance of Firmicutes ranged from 40.34% to 68.82%. Bacteroidetes were then the second predominant bacterium, and their abundance ranged from 26.36% to 40.81%. Overall, the relative abundance of Firmicutes tended to increase as pigs aged, however, of Bacteroidetes decreased.
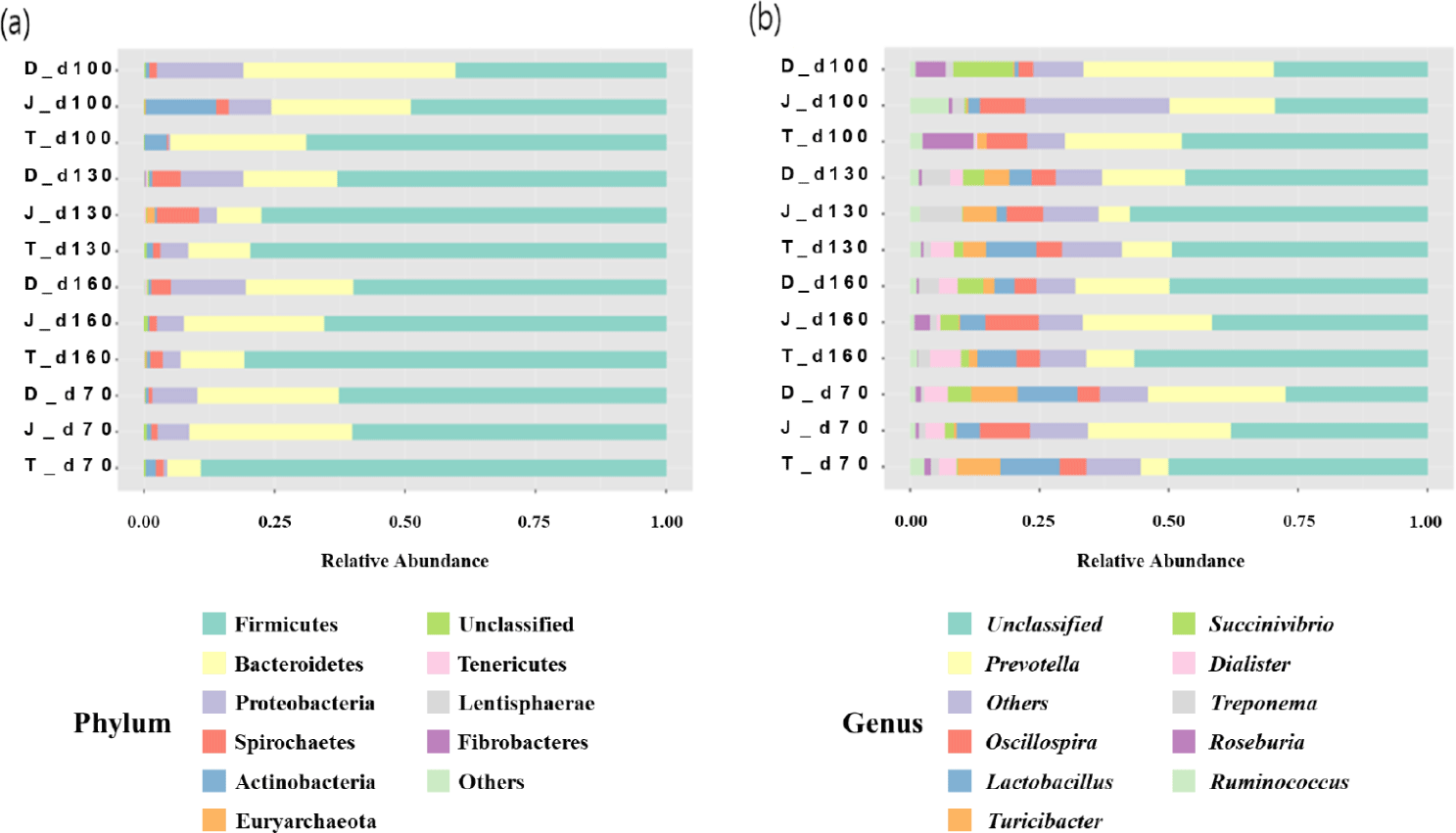
The relative abundances of different bacterial taxa at the genus level among the three groups at 70, 100, 130, and 160 days of age is shown in Fig. 4B. Prevotella was one of the most abundant genera regardless of age of the pigs.
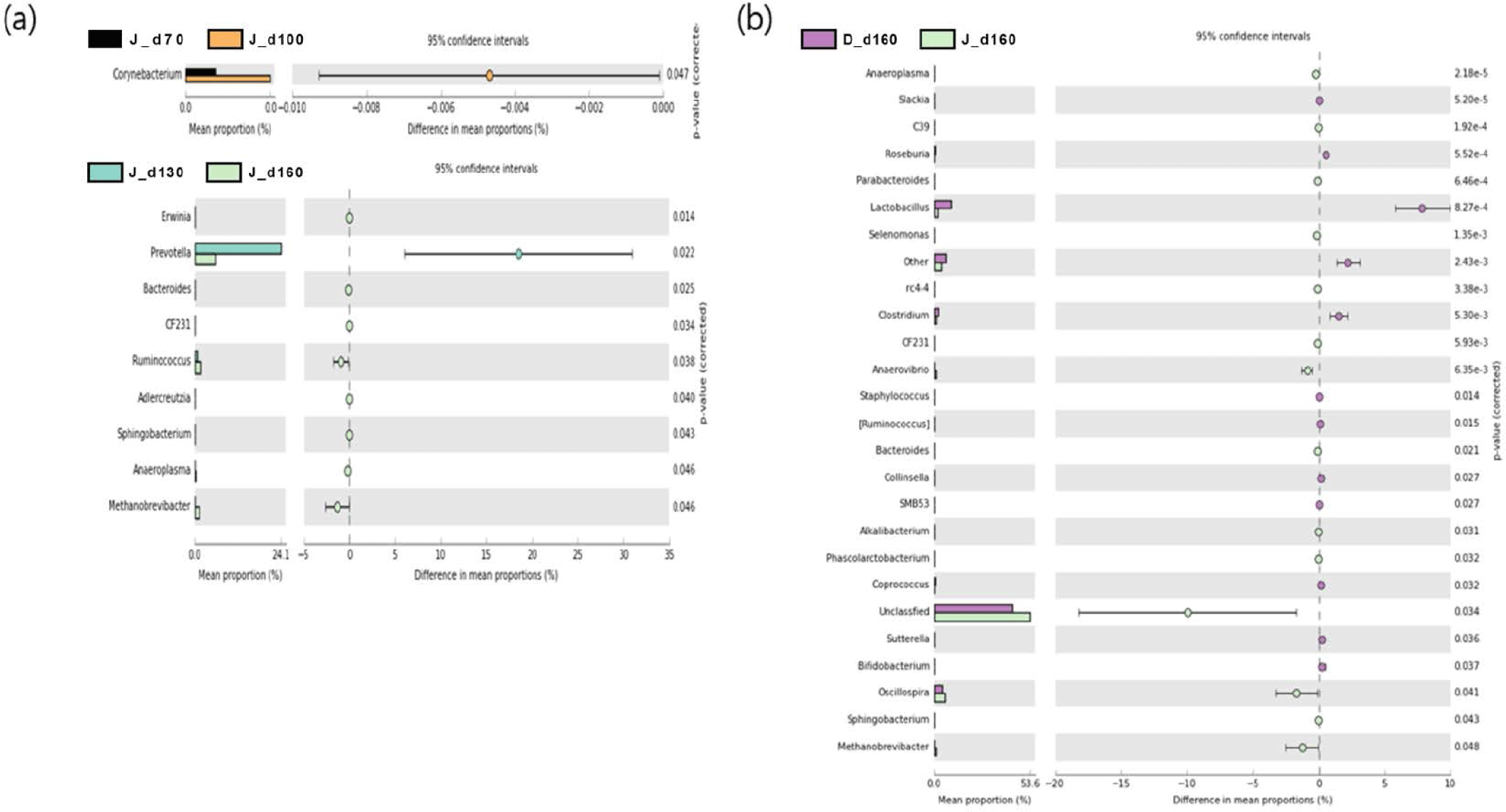
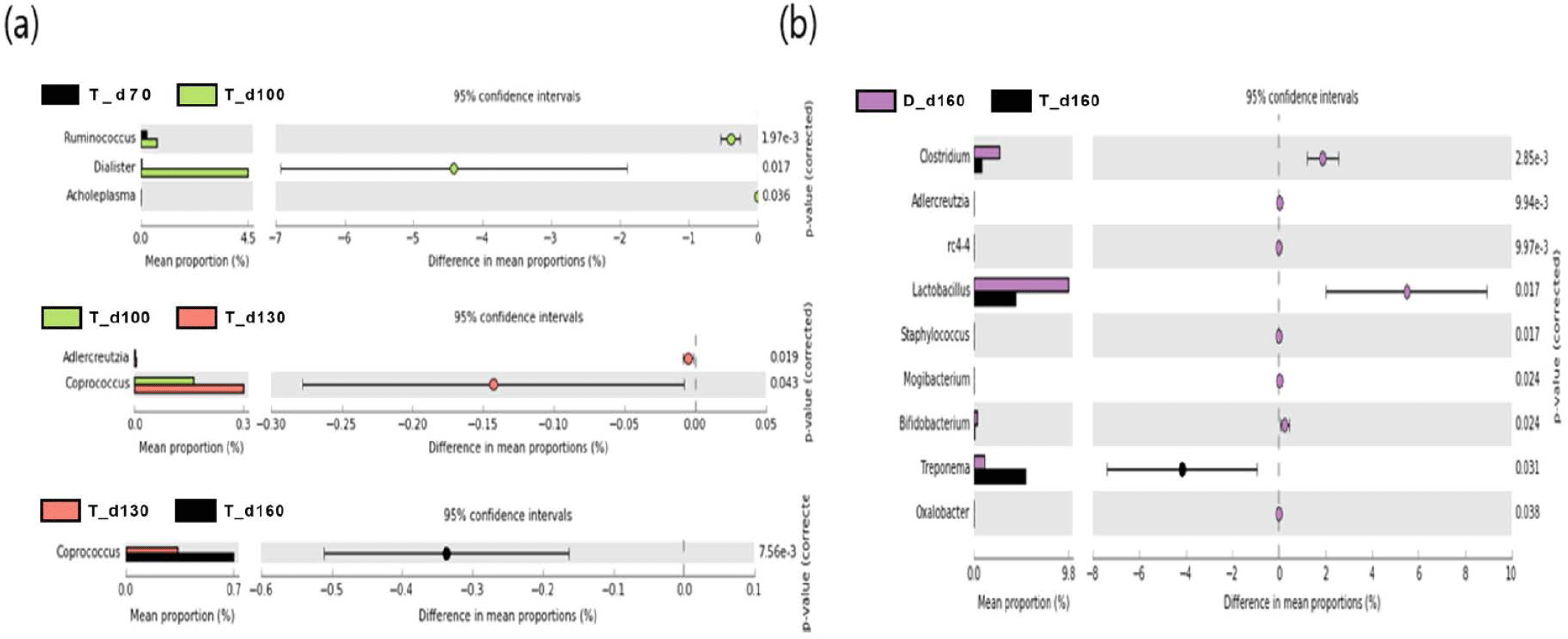
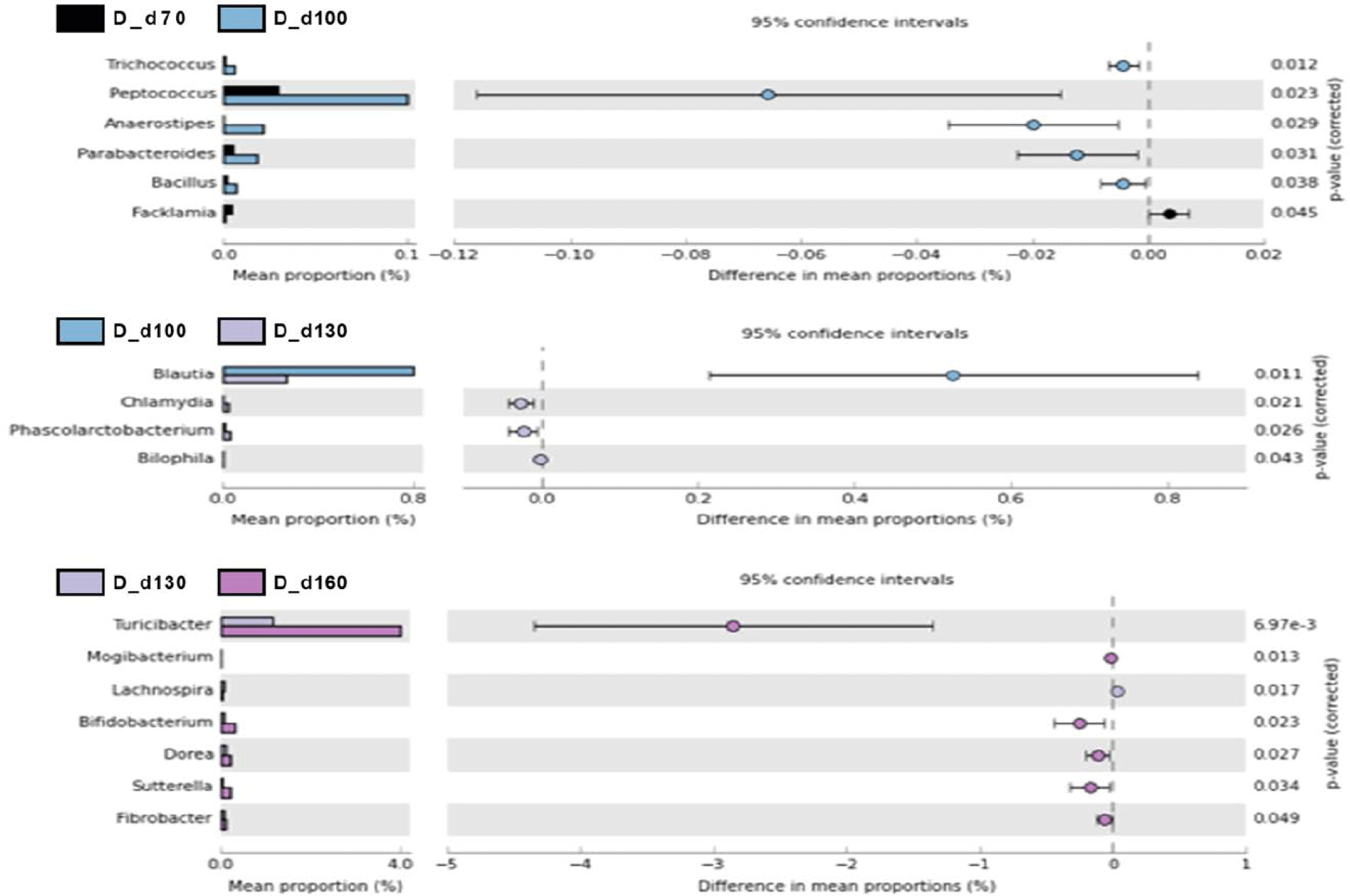
We used a two-sided Welch’s t-test in STAMP to compare the relative abundances of taxa at the genus level at different ages, and they were visualized using an extended error bar plot. In group J, the comparison of bacterial communities between 70 and 100 days of age showed that the relative abundance of Corynebacterium significantly increased at 100 days of age (p < 0.05) (Fig. 5A). The comparison of bacterial communities between 130 and 160 days of age showed that the relative abundances of genus Erwinia, Bacteroides, CF231, Ruminococcus, Adlercreutzia, Sphingobacterium, Anaeroplasma, and Methanobrevibacter were significantly higher at 160 days of age, while the relative abundance of Prevotella was significantly higher at 130 days of age (p < 0.05) (Fig. 5A). The comparison of the relative abundances of genera at 160 days of age between D and J groups showed that the relative abundances of Anaeroplasma, C39, Parabacteroides, Selenomonas, rc4-4, CF231, Anaerovibrio, Bacteroides, Alkalibacterium, Phascolarctobacterium, Unclassfied, Oscillospira, Sphingobacterium, and Methanobrevibacter were significantly higher in group J than those of group D (p < 0.05). However, the relative abundances of genus Slackia, Roseburia, Lactobacillus, Clostridium, Staphylococcus, (Ruminococcus), Collinsella, SMB53, Coprococcus, Sutterella, and Bifidobacterium were significantly higher in group D in comparison to group J (p < 0.05) (Fig. 5B). In group T, the comparison of bacterial communities between 70 and 100 days of age showed that Ruminococcus, Dialister, and Acholeplasma were significantly higher in pigs of 100 days of age (p < 0.05). The relative abundance of Coprococcus increased as pigs aged (p < 0.05) (Fig. 6A). Comparison of microbial communities between groups D and T at 160 days of age showed that the relative abundances of Clostridium, Adlercreutzia, rc4-4, Lactobacillus, Staphylococcus, Mogibacterium, Bifidobacterium, and Oxalobacter were significantly higher in group D than those of group T (p < 0.05), however, the relative abundance of Treponema in group D was significantly lower than group T (p < 0.05) (Fig. 6B). In group D, the relative abundances of Trichococcus, Peptococcus, Anaerostipes, Parabacteroides, and Bacillus were significantly higher at 100 days of age compared to those of 70 days of age (p < 0.05). From 100 to 130 days of age, the relative abundances of Chlamydia, Phascolarctobacterium, and Bilophila increased while the relative abundance of Blautia significantly decreased (p < 0.05). From 130 days to 160 days of age, the relative abundances of Turicibacter, Mogibacterium, Bifidobacterium, Dorea, Sutterella, and Fibrobacter significantly increased, whereas that of Lachnospira significantly decreased (p < 0.05) (Fig. 7).
DISCUSSION
This study was performed to evaluate the relationship between the gut microbiome and the growth performance of the pigs from three different farms. The average age of pigs in three different farms when they reached the average market weight of 118 kg were < 180 days, about 190 days and > 200 days, respectively. Beta-diversity was assessed using both weighted and unweighted UniFrac distance. Weighted UniFrac distance accounts for the relative abundance of OTUs whereas unweighted UniFrac distance accounts for only community membership i.e. presence or absence of OTUs [10]. The results of beta-diversity showed no significant separation of microbial community among the groups. Alpha diversity analysis showed increase in bacterial diversity as pigs aged, suggesting significant changes in measures of species uniformity and species abundance in pigs. However, there was no significant differences observed among groups in Chao1, the number of observed OTUs, and the Shannon index.
The gut microbiome of all three groups showed to be dominated by the phyla Firmicutes and Bacteroidetes, being consistent with previous studies [11–13]. It was confirmed that the relative abundances of Firmicutes and Bacteroidetes were significantly different among the groups. In this experiment, the breed and nutritional level of the pigs were similar, but the environmental management program was different for each farm. These results are consistent with previous studies showing changes in microbial flora influenced by environmental management [14,15]. One of the most interesting observations in this study was higher relative abundances of Bifidobacterium, Clostridium and Lactobacillus at the genus level in group D. Pigs do have Bifidobacterium spp. as their major component of intestinal microbiota, however the amount is less than Lactobacillus spp., as determined by both culture-dependent [16] and culture-independent methods [17]. A prior study established positive impacts of Lactobacillus on feed efficiency of crossbred pigs (Duroc × [Landrace × Yorkshire]) [18]. Some Clostridium species have also been demonstrated to modulate the colonic luminal metabolome through production of short-chain fatty acids like butyrate, which aids in maintaining the gut health [19,20]. So far, very few studies have evaluated the relationship between the pig’s growth performance and their gut flora.
In this study, we evaluated the gut microflora at different stages of growth, and the results confirmed microbial shifts as pigs aged. Our results will be useful for designing host-microbial interaction studies, especially in the pig industry, for promoting overall health and growth in pigs.