INTRODUCTION
Antibiotics have been used in livestock production for a longtime [1]. They have been employed in intensive farming to boost productivity. In animals, antibiotics is commonly used for not only treatment but also for increasing the effectiveness of animal feed, promoting animal growth, and preventing diseases at a time when their immunity is low due to stress [2]. However, careless usage of antimicrobials lead to a rise in antibiotic-resistant bacteria and genes [3,4]. Increased antibiotic resistance can raise disease incidence and cause chronic health problems in livestock. So, the use of antibiotics for fostering growth in livestock is no longer permitted in the EU and South Korea [5].
Antibiotic-resistant bacteria can be discharged into the surrounding environment, the soil and water which can act as sources for transmission of antibiotic-resistant bacteria [2]. In addition, there are reports of transmission of antibiotic-resistant genes in a surrounding environment adjacent to the pig farm [6]. During the weaning period, piglets are exposed to a variety of stressors, such as changes in feed composition and environment [7,8]. Prior researches have shown that these stressors alters the piglet gut microbiome during nursing and weaning phases [9]. However, it is uncertain whether the gut microbial shifts are associated with the increased antibiotic-resistant genes in pigs during the weaning transition. In addition, from pre-weaning to weaning pigs are gradually exposed to more environment such as soil and water. Due to such environmental exposure, antibiotic resistance in piglets can increase and remain in animal-derived products for human consumption, so antibiotic-resistant gene is observed as “One Health subject” [2]. Therefore, the whole metagenome shotgun sequencing was used in this study to evaluate changes in microbiome and antibiotic-resistant genes during weaning transition.
MATERIALS AND METHODS
Fecal samples were collected from 8 piglets in a gap of 1 week, firstly just before weaning (21 d of age) and secondly one week after weaning (28 d of age) and placed in sterile test tubes and stored at −80℃. After weaning, the piglets were fed a conventional nursery feed based on soybean meal and corn, that complied with National Research Council standards [10,11] of nutrient requirements of weaned piglets. The piglets had unrestricted access to feed and water. The piglets received no additional supplements or antibiotics through the whole duration of the experiment.
200 mg of feces per sample were used for the total DNA extraction, using QIAamp Fast DNA Stool Mini Kit (QIAGEN, Hilden, Germany), as per the instructions provided by the manufacturer. Cell lysis was achieved by bead-beating the samples twice for 2 min at 300 rpm, followed by a 5-minute incubation period in a water bath at 70℃ between beatings. A Colibri Microvolume Spectrometer (Titertek Berthold, Pforzheim, Germany) was used to measure the concentrations of DNA and only the samples that had OD260/280 ratio between 1.80 and 2.15 underwent further processing.
The paired-end shotgun sequencing using the Illumina Hi-Seq 2000 platform was used to sequence the extracted DNA representing the fecal microbial communities. The whole metagenome shotgun sequencing was carried out at Macrogen (Seoul, Korea).
Whole metagenome shotgun sequencing was carried out on a subset of eight samples randomly chosen (four samples from the same piglets at 21 and 28 days of age) to examine the microbial diversities and fecal microbial functions present in the piglet fecal samples. For the microbial functional analysis, the obtained raw sequence data in FASTQ format were imported to Metagenomics Rapid Annotation using Subsystem Technology (MG-RAST) pipeline. All the classified subsystem reads were normalized in MG-RAST. To account for differences in sequencing depth of samples, DESeq was utilized within the analysis pipeline [12]. MG-RAST pipeline was used for removing artificial duplicate reads [13]. MG-RAST also removed sequence reads that matched the host’s genome through Bowtie. [14]. The Reference Swine Genome (Sus scrofa, NCBI v10.2) available in MG-RAST was used to filter out the host-derived metagenomic reads [13]. The SEED Subsystems database, which is a collection of functionally related protein families, was used for the functional annotation of the sequence reads [15]. Using an e-value of less than 1 × 10−5, minimum identity of 60%, and a minimum alignment length of 15 amino acids for protein, the similarity search between sequence reads and the SEED databases was performed. For the taxonomic assignment of the sequences, the Greengenes reference database was utilized. Significant variations in functional profiles and taxonomic compositions between the nursing and weaned pigs were determined based on Multiple t-test, using STAMP and GraphPad Prism version 7.00 (GraphPad Software, La Jolla, CA, USA).
RESULTS AND DISCUSSION
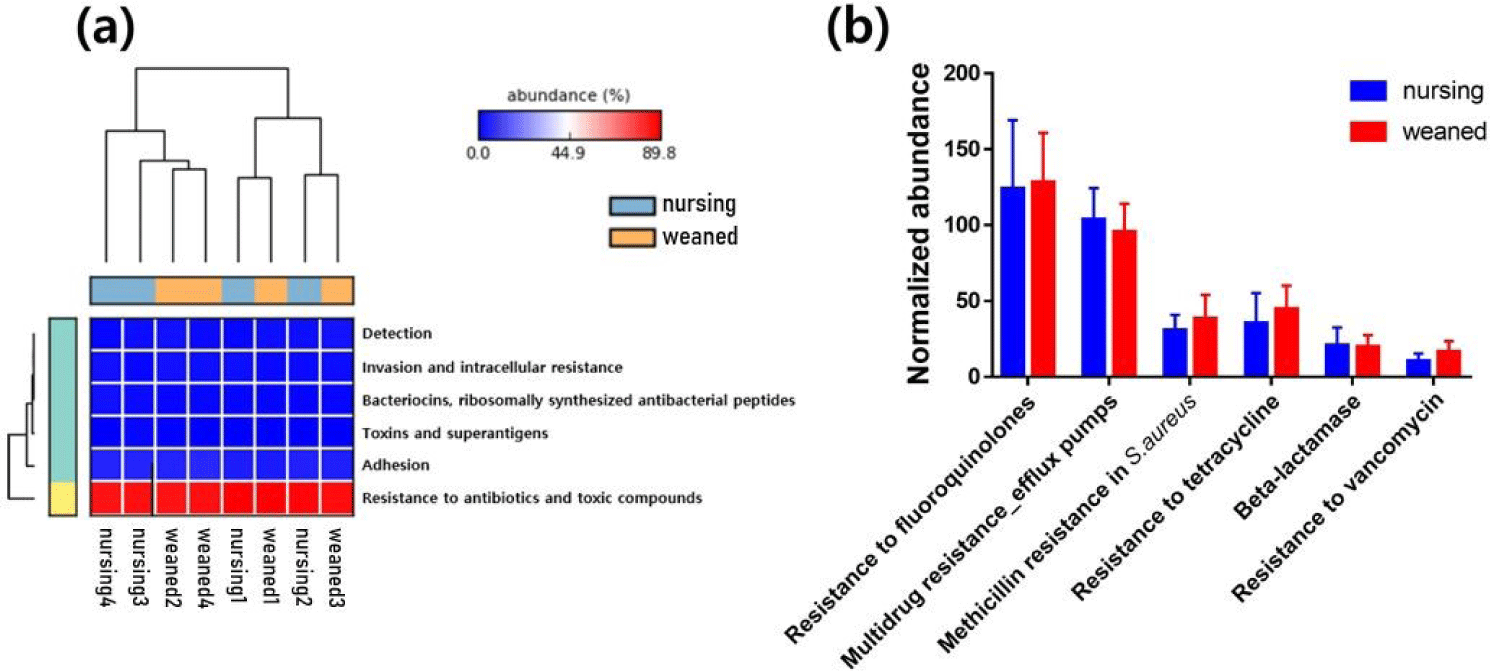
In general, a total of 50,440,732 sequences was obtained by the whole metagenome shotgun sequencing using HiSeq Illumina platform. After microbial functional analysis using MG-RAST pipeline, 28 level 1 SEED subsystems were identified in both nursing and weaned piglet metagenome, and the functional gene groups associated with level 2 SEED subsystem “Resistance to antibiotics and toxic compounds” within the level 1 SEED subsystem “Virulence, disease, and defense” (Fig. 1A) were studied further. At the level 3 SEED subsystems within the level 2 SEED subsystem “Resistance to antibiotics and toxic compounds”, gene families related to “Resistance to vancomycin”, “Resistance to tetracycline”, “Multidrug resistance_efflux pumps” and “Methicillin resistance in Staphylococcus aureus(MRSA)” tended to increase, although not significantly in the weaned piglets than nursing ones (Fig. 1B). In the “Resistance to vancomycin”, the vancomycin binding blocking protein “B-type resistance protein VanW” was significantly enriched (p < 0.05), and “response regulator VanR” and “Sensor histidine kinase VanS (EC 2.7.3.-) tended to increase (Fig. 2A). In “Resistance to tetracycline”, only ribosomal protection proteins (RPPs) related genes were detected and increased on average (Fig. 2B). In the “Multidrug resistance_efflux pumps”, “Multi antimicrobial extrusion protein MATE family of MDR efflux pumps” and “RND efflux membrane fusion protein” that related in extrusion of toxic substrates into the cellular environment were largely present in the weaned piglets (Fig. 2C). In the “MRSA, “UDP-N-acetylmuramoylalanyl-D-glutamate--2, 6-diaminopimelate ligase (murE)”, “D-alanyl transfer protein DltB”, “Poly D-alanine transfer protein DltD”, “Methicillin resistance and cell wall biosynthesis protein FmtB” and “D-alanine-poly ligase subunit 2 (DltC) (EC 6.1.1.13)” which function as methicillin resistance and cell wall biosynthesis were slightly higher in the weaned piglets than nursing piglets (Fig. 2D).
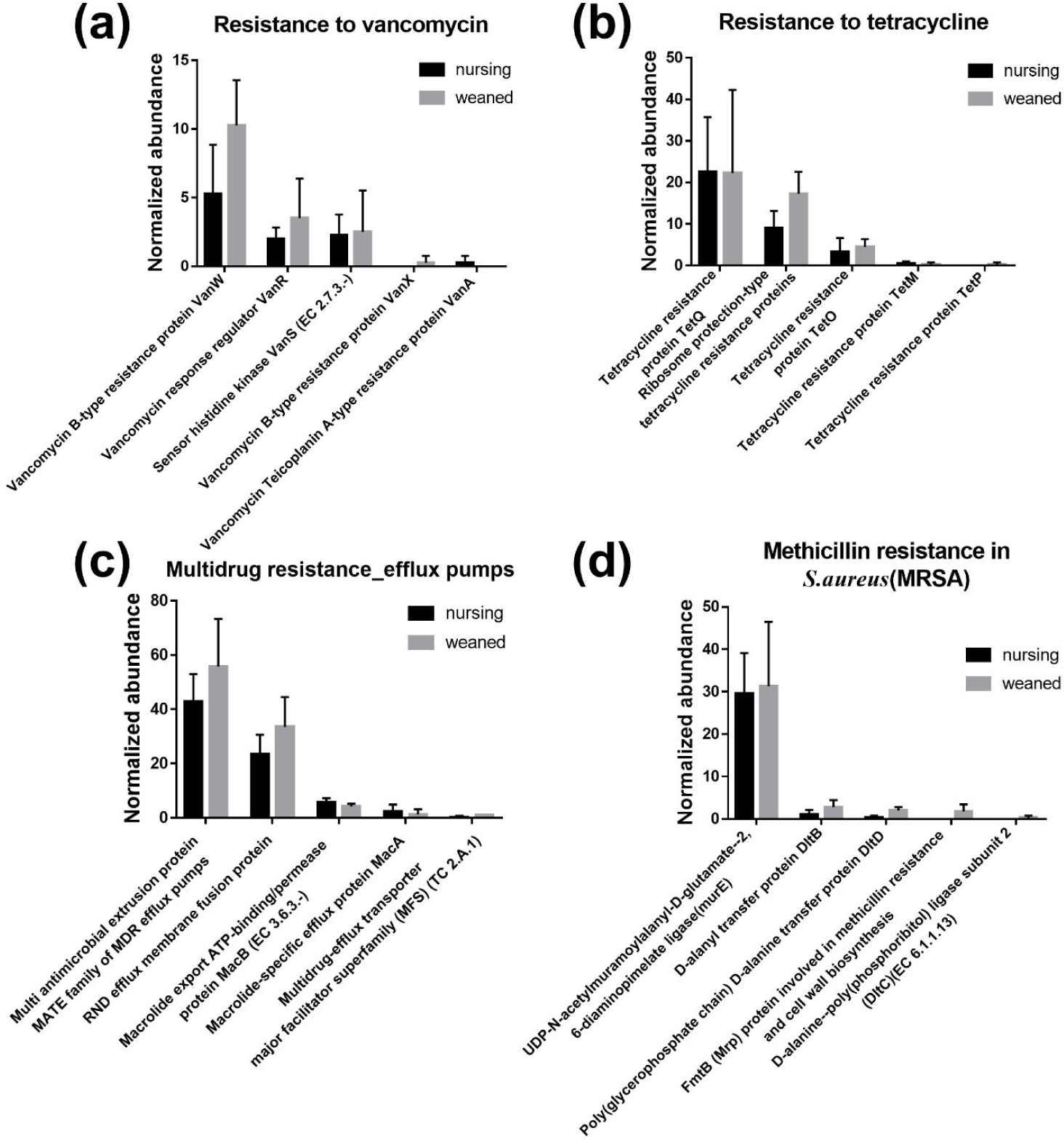
Vancomycin is a β-lactam glycopeptide antibiotics that binds to acyl-D-ala-D-ala of the peptidoglycan cell wall to inhibit cell wall biosynthesis, but acts only on gram-positive bacteria because it cannot pass through the lipid bilayer of gram-negative bacteria [16]. There are two types of glycopeptide resistance through conversion to D-ala-D-lac [17,18]. As a results of detection, B-type proteins were higher which is resistance to vancomycin and vulnerable to teicoplanin. Tetracycline prevents the binding of aminoacyl-tRNA and ribosome with 30S prokaryotic ribosomal subunit as a target, resulting in translation inhibition [19,20]. Mainly, TetQ is found in anaerobic gram-negative species with a limited host range, TetM is located in specific part of Enterococcus faecalisand TetO is found in Gram-positive species such as Streptococcus pyogenes[21–23]. These RPPs restore A-site delivery of aminoacyl-tRNA to give resistance to tetracycline [24]. According to sources of energy utilized (adenosine triphosphate [ATP] or Hydrogen/sodium ions) and amino acid sequences, efflux pumps are classified into five major active super families: Major facilitator superfamily (MFS), Resistance nodulation cell division superfamily (RND), MATE, ATP Binding cassette transporter (ABC) and Small multidrug resistance (SMR) [25]. All bacteria contain several efflux pumps, which extrude not only antibiotic substrates but also non-antibiotic substrates such as heavy metals, dyes, detergents and organic solvents [26–28]. There are reports of fluoroquinolone transported by MATE in mammals, and RNDs were distributed in gram-negative bacteria to export β-lactams out of outer-membrane [29–33]. The murE and fmtB gene detected in MRSA are associated with cell-wall peptidoglycan biosynthesis, and nothing else is known about the fmtB gene [34,35]. The lipoteichoic acid, which constitutes the cell wall of gram-positive bacteria, is made through D-alanylation of LTA through DltA-D [36]. Due to the mechanism of methicillin, which binds to PBP2a to biosynthesis of cell wall, it is considered that methicillin resistance increased as the number of genes associated with cell wall biosynthesis increased.
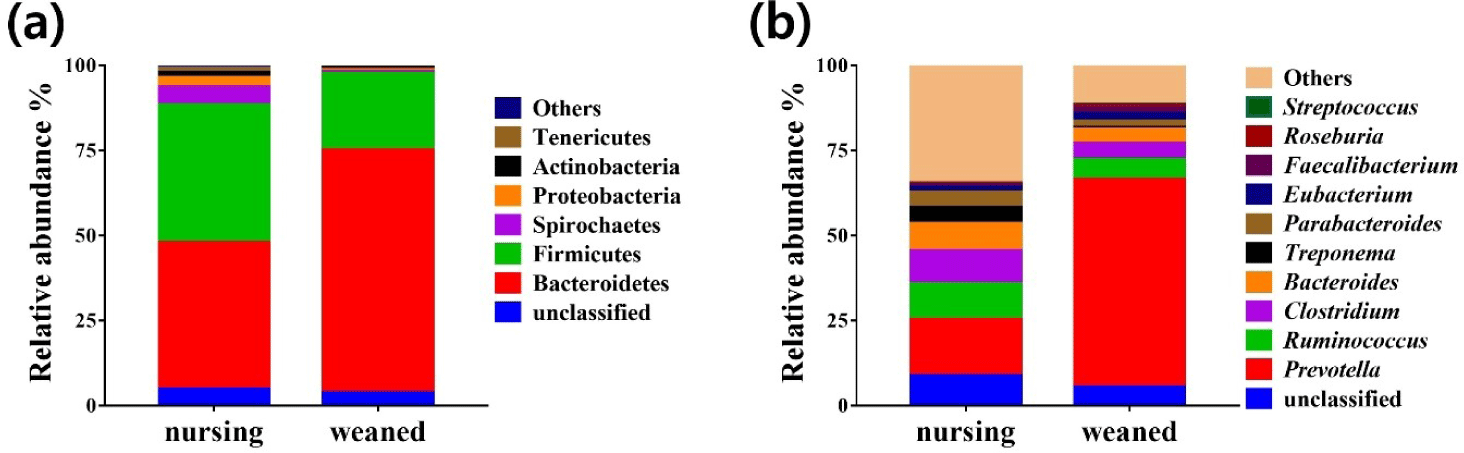
Using the whole metagenome shotgun sequences, the relative abundance of the gut microbiota was compared at the phylum and genus levels during the weaning transition (Fig. 3). The results from total sequence analysis confirmed significant change in bacterial composition after the weaning transition. At the phylum level, nursing piglet showed presence of Bacteroidetes (43.18%), Firmicutes (40.5%), Spirochaetes (5.21%), Proteobacteria (2.8%), Actinobacteria (1.47%), Tenericutes (1.2%) and other 6 phylum (0.8%) (Fig. 3A). After weaning transition, piglet gut microbiota were consisted of Bacteroidetes (71.5%), Firmicutes (22.46%), Spirochaetes (0.57%), Proteobacteria (0.43%), Actinobacteria (0.71%), Tenericutes (0.28%) and other phylum (0.26%) (Fig. 3A). Compared to before the weaning transition, Bacteroidetes increased significantly from 40.5% to 71.5% on average (p < 0.01). At the genus level, Prevotella showed the largest amount of abundance, and enriched on average 16.57% to 61.09% that significantly increased after weaning (p < 0.001). Streptococcus increased only by a small amount from 0.13% to 0.28%. The relative abundances of other genera were shown in Fig. 3B. The genus Prevotella, which has the ability to break down protein and carbohydrate in feed, increased significantly with the change in diet from milk to grain feed during the weaning transition [37]. Unlike the tendency of antibiotic resistance genes to increase after weaning, there were no significant increase in the relative abundance of major antibiotic resistant bacteria such as Staphylococcus aureus, Enterococcus and Mycobacterium tuberculosis. The data from this study showed that changes in diet and environment can be a potential health risk factor and may affect piglets.