
INTRODUCTION
In many feed systems, methionine (Met) is known as a limiting amino acid (AA) for milk protein synthesis in dairy cows [1-3]. Met plays a critical role in protein synthesis and is active in many other biochemical and cellular processes [2,3]. The primary role of Met is to initiate mRNA translation by binding methionyl-tRNA into the 40S ribosome, then joining with the 60S ribosome to form the 80S ribosome [4]. Met supplementation has been reported to increase protein expression, mammalian target of rapamycin (mTOR) phosphorylation, and beta-casein expression [5]. Supplementation with Met in bovine mammary epithelial cells causes the increase of mTOR pathway activation and phosphorylation of glycogen synthase kinase 3 which acts as an inhibitor of mTOR [6]. Met supplementation in bovine mammary epithelial resulted in positive effects on milk protein synthesis because of phosphorylation of proteins related to the mTOR pathway [7]. Met infusion into bovine mammary tissue has also been reported to increase S6 kinase beta-1 (S6K1) and ribosomal protein (RP)S6 phosphorylation [8]. Several past studies have shown that nutrient–gene interaction allows Met to directly regulate the expression of genes linked to milk protein synthesis [9,10].
D-Met is one form of Met that can be made synthetically and used as a supplement in the diets of dairy cows. The roles of enzymes are particular to the specific isomer. D-Met is converted into L-form via a two-step process, mainly in the liver, kidney, and other tissues [11-13]. Amino acid (AA) oxidase removes the amine group via an oxidation reaction to form the intermediate 2-keto-4-methylthiobutanoic, followed by attachment of transaminase to the amine group forming the L-Met [11-13].
The Met hydroxy analog 2-hydroxy-4-methylthiobutanoic acid (HMBi), a Met pre-cursor, has long been proposed to provide Met and thus increase the milk protein contents in dairy cows fed with Met-limited ration [14]. The chemical structure of HMBi is mainly similar to that of Met. The main difference in their structures is that instead of an amine group, HMBi has a hydroxyl group (OH). The analog also exists in both D- and L- isomer forms. HMBi has several beneficial effects on rumen fermentation, including enhancing cellulolytic activity, fiber degradation, and increased acetate production [15-17].
Recently, different omics studies detected distinct clusters of differentially concentrated metabolites following treatment with various ratios of Met, Lysine, Phenylalanine, Valine, and Tryptophan [18-20]. However, to our knowledge, no studies have applied proteomic and metabolomic approaches to analyze the effects of supplementation with D-Met and its precursor HMBi on the beta-casein synthesis and milk protein synthesis-related pathways. The present study aims to determine the effect of adding D-Met and the Met precursor HMBi on the stimulation of milk protein synthesis in bovine mammary epithelial cells. It also investigates the related pathways that increase protein synthesis and beta-casein expression in immortalized bovine mammary epithelial cell lin (MAC-T) cells using proteomic and metabolomic data.
MATERIALS AND METHODS
MAC-T cells which are immortalized mammary epithelial [21] came from McGill University, Canada, and were cultured following the procedure of Wang et al. [21,22]. The AA profile of the differentiation medium is shown in Table 1. Cell confluency can be estimated by considering the doubling time characteristics of our cells.
An initial experiment was done to determine the effects of various dosages of supplemental Met and its culture time on beta-casein mRNA expression. Additional Met (+0.3, +0.6, +0.9 mM) were supplemented to lactogenic medium without fetal bovine serum. Cells were harvested 24 hours.
Beta-casein secretion on MAC-T cells was harvested at 0, 12, 24, 36, 48, 72, 96, and 120 h after incubating with the lactogenic medium to determine the effect of the treatments on culture time. Preliminary study shows that 0.6 mM and incubation time at 72 h were the best dosage and incubation time respectively and were used uniformly for all treatments. D-Met and HMBi were obtained from Sigma-Aldrich (M9375) (Sigma-Aldrich, St. Louis, MO, USA) and Adisseo MetaSmart® (Adisseo, Antony, France), respectively. Treatments were replicated six times and randomly assigned on a 6-well plate.
Using TRIzol reagent (Life Technologies, Carlsbad, CA, USA), the total RNA from MAC-T cells were extracted. The RNA quality and quantity were determined in NanoDrop 1000 spectrophotometer using RNA-40 module (Thermo Fisher, Waltham, MA, USA). Values of > 1.8 A260/280 were acceptable. The cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA). The sample was incubated first at 25°C for 5 min, then 42°C for 30 min, and finally at 85°C for 5 min. The cDNA quantity and quality were evaluated using the ssDNA-33 module of the Thermo NanoDrop 1000 spectrophotometer (Thermo Fisher).
RT-PCR was done with a total volume of 20 μL/well using 96-micro-well plates (Sigma-Aldrich). Cycling temperature was performed using the T100 Thermal Cycler System (Bio-Rad). Beta-actin served as the reference gene. The reaction mixture is shown in Table 2. RT-PCR oligonucleotide sequences of forward and reverse primers specific for the target genes that were tested and validated are shown in Table 3. RT-PCR reactions were found in Table 4. The Livak method was used to determine the relative gene expression of the target gene to the control group using the threshold cycle (2–ΔΔCT method) [23] to analyze relative gene expression changes from real-time quantitative PCR experiments. Relative quantification of expression levels of target genes in the treatment group was compared to the untreated group. A Gene expression ratio equal to 1 was used for all the control values. Therefore, a relative gene expression value greater or less than 1 correspond to a fold increase or decrease in expression relative to the control. Furthermore, to test the significance of the differences observed, least significant difference (LSD) test was performed.
| Component | Amount |
|---|---|
| cDNA | 50 ng |
| Forward primer | 0.6 μL |
| Reverse primer | 0.6 μL |
| PCR master mix | 10 μL |
| (DEPC)-treated water | 6.3 μL |
| PCR cycling step | Temperature | Time | Cycle |
|---|---|---|---|
| Initial incubation | 95°C | 3 min | 1 |
| Denaturation | 95°C | 10 sec | 50 |
| Annealing | 55°C | 10 sec | 1 |
| Extension | 72°C | 30 sec | 1 |
The melting curve was also considered to determine the Ct. This is to keep the cycle threshold firmly within the geometric phase. This is very important to calculate the fold changes from sample to sample and to get quantity information from a standard curve. Certain regions of the melting curve were also avoided, specifically, Ct was set not to be too low to avoid the noise during amplification. Also, too high Ct was also avoided wherein it is already linear or in the plateau phase of amplification where data are less predictable. We target the spot where all the curves are straight and parallel to one another which is the point where the precision of our replicates is the highest. It is somewhere toward the middle of the geometric phase or maybe slightly higher.
After 72 h of incubation, the culture medium was collected for protein quantification by centrifuging at 300×g for 5 min at 4°C by removing the dead cell fragments. The medium was quantified using a Pierce bicinchoninic acid assay (BCA) kit (Pierce Biotechnology, Rockford, IL, USA). The cells attached at the bottom of the well were then washed with 1× phosphate-buffered saline (PBS). Then 200 μL cell lysis buffer (pH 8.3) containing 10 mM Tris/HCl, 8 M urea, 5 mM ethylenediaminetetraacetic acid (EDTA), 4% 3-[(3-cholamidopropyl) dimethylammonio]-1-propane sulfonate (CHAPS), and 1× protease inhibitor cocktail (GE Healthcare, Piscataway, NJ, USA) were added. Cell lysates were stood at 20°C for 30 min and centrifuged at 14,000×g for 30 min at 20°C [21]. The collected cells underwent cell protein quantification by BCA using the Pierce Protein Assay Kit (Pierce Biotechnology).
Lysis buffer containing 20 mM Tris, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.1% sodium dodecyl sulfate (SDS), and complete EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland) was used to extract cell proteins. Cell lysates were incubated at 4°C for 30 min and then centrifuged at 13,000×g for 10 min at 4°C [24]. All treatments were replicated three times.
The proteome analysis was performed according to the procedures described by Ishihama et al. [24]. The exponentially modified protein abundance index (emPAI)-based abundances compared with the actual values were within 63% on average, which is similar or better than the determination of abundance by protein staining [24]. Also, metabolite quantity determination was done as described by a previous study done by Park et al. [25].
Statistical analyses were done using SAS v. 9.1 software (SAS Institute, Cary, NC, USA). The dose responses were analyzed by computing the polynomial orthogonal contrasts (linear, quadratic, cubic) of responses to the supplementation of Met. Data were analyzed by two-way analysis of variance (ANOVA) with supplement type and plate as fixed effects. Significant differences among treatments were evaluated using LSD test. The significant difference was determined at a level of p < 0.05. The experimental model was:
Where: μ = grand mean
αi = effect of Met type and the Met precursor HMBi
βj = effect of plate
γij = interaction of treatment (D-Met/HMBi) and plate
εijk= error variability
Significant increases and decreases in protein quantity were determined by the semi-quantification relative ratio (the ratio was > 2 or > 0.5 respectively) [26,27]. The detected proteins were analyzed for pathway analysis (Bos taurus) by using a website program (http://www.pantherdb.org accessed on 26 October 2021). Differentially expressed metabolites and affected pathway detection metabolites were analyzed using Dunnett’s Multiple Comparison test by SAS software (V 9.1, SAS Institute). Pathway detection metabolites affected by the application of treatments were analyzed using the Metabo Analyst 3.0 program (University of California, Davis), a pathway detection metabolite tool for pathway analysis of cattle (Bos taurus).
RESULTS AND DISCUSSION
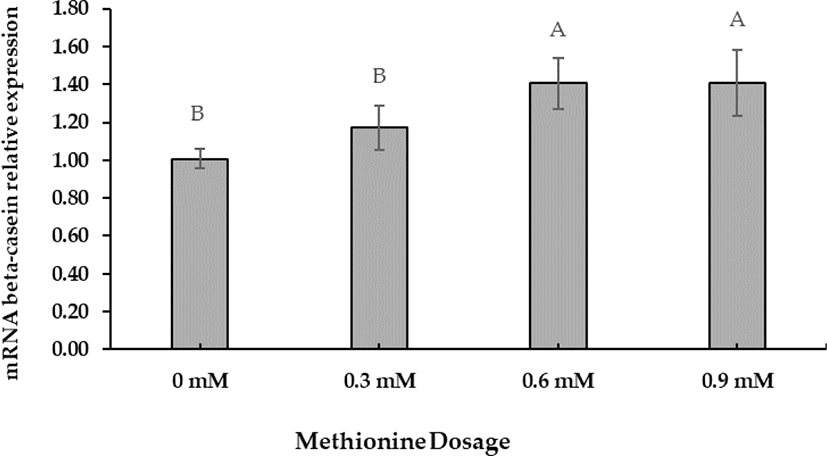
The preliminary Met dosage test showed that +0.6 mM was the optimal concentration for increasing beta-casein mRNA gene expression. Beta-casein mRNA expression increased at a Met-dose of +0.6 mM (Fig. 1). Polynomial orthogonal contrasts (linear, quadratic, cubic) analyzed the dose responses to Met supplementation (Table 5). Linear contrasts of dose responses revealed significant differences (p < 0.05).
| Contrast | DF | Contrast SS | Mean square | F-value | Pr > F |
|---|---|---|---|---|---|
| Linear | 1 | 0.619 | 0.619 | 6.20 | 0.022 |
| Quadratic | 1 | 0.039 | 0.039 | 0.39 | 0.538 |
| Cubic | 1 | 0.027 | 0.027 | 0.27 | 0.608 |
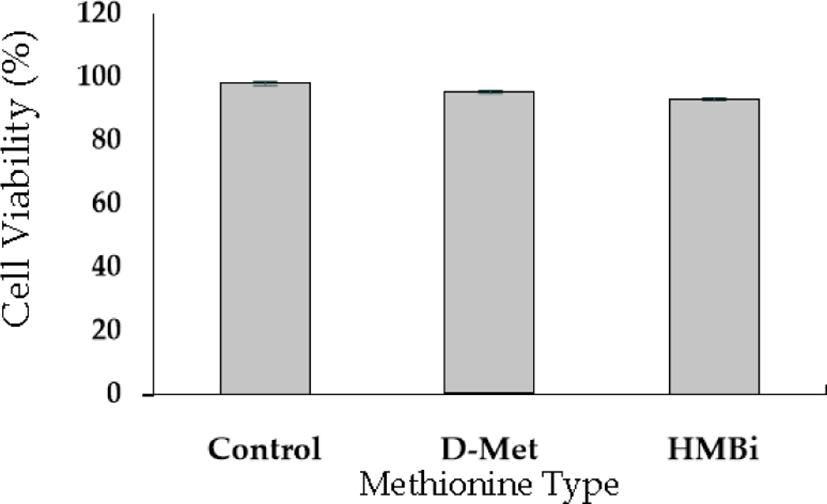
A cell viability test was performed to evaluate D-Met isomer and HMBi supplementation; the 0.6-mM dose had no significant toxic effect than the control (Fig. 2). The result implies that +0.6 mM is the most effective level for increasing beta-casein mRNA expression identical to the previously reported result [27-29].
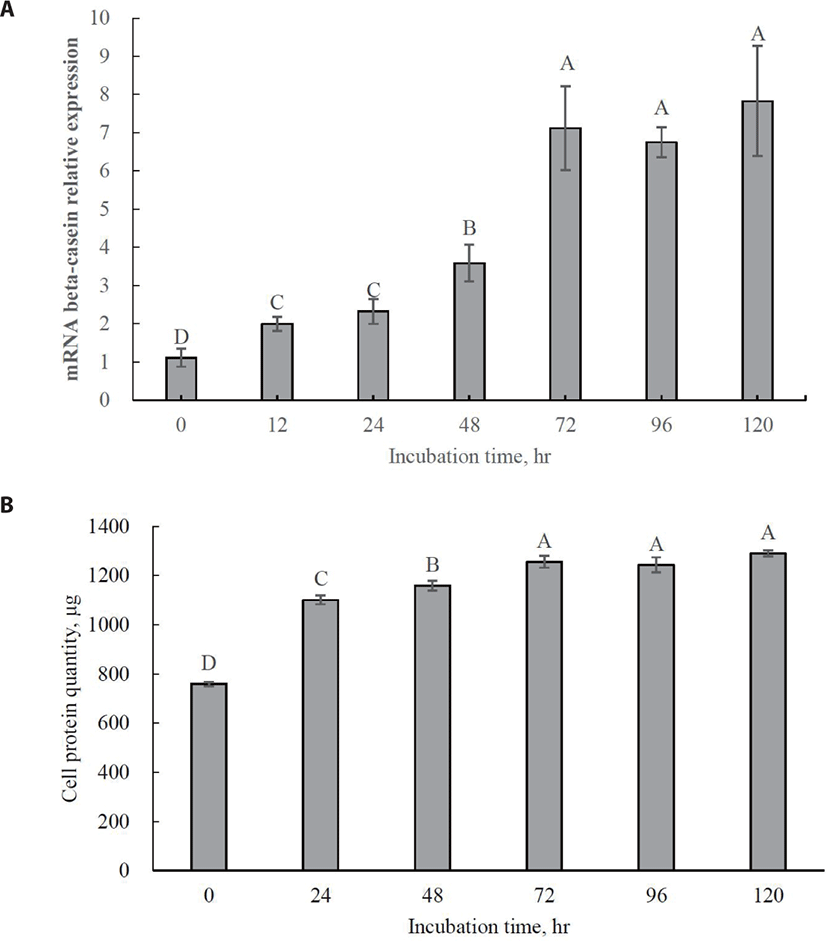
The optimal incubation time for increasing beta-casein mRNA expression was 72 h (Fig. 3A; p < 0.05). No significant increase in beta-casein mRNA expression was observed even after 72 h. Maximum cell protein synthesis coincides with maximum beta-casein mRNA expression (Fig. 3B; p < 0.05). This result indicates that 72 h should be used as the optimal incubation time for further tests of Met isomer and precursor efficacy since MAC-T cells were completely differentiated into beta-casein-secreting cells. Previous studies have reported that relative gene expression for beta-casein production is generally increased by supplementation with Met [28,29].
No significant difference in cell protein upon addition of the treatments (Fig. 4A; p > 0.05). The culture medium treated with HMBi led to significantly higher protein content than the control, yet it was not significantly different from the D-Met treatment (Fig. 4B; p > 0.05). Availability of Met is critical for regulating translation and a major limiting factor for protein synthesis in the mammary epithelial cells [30-32].
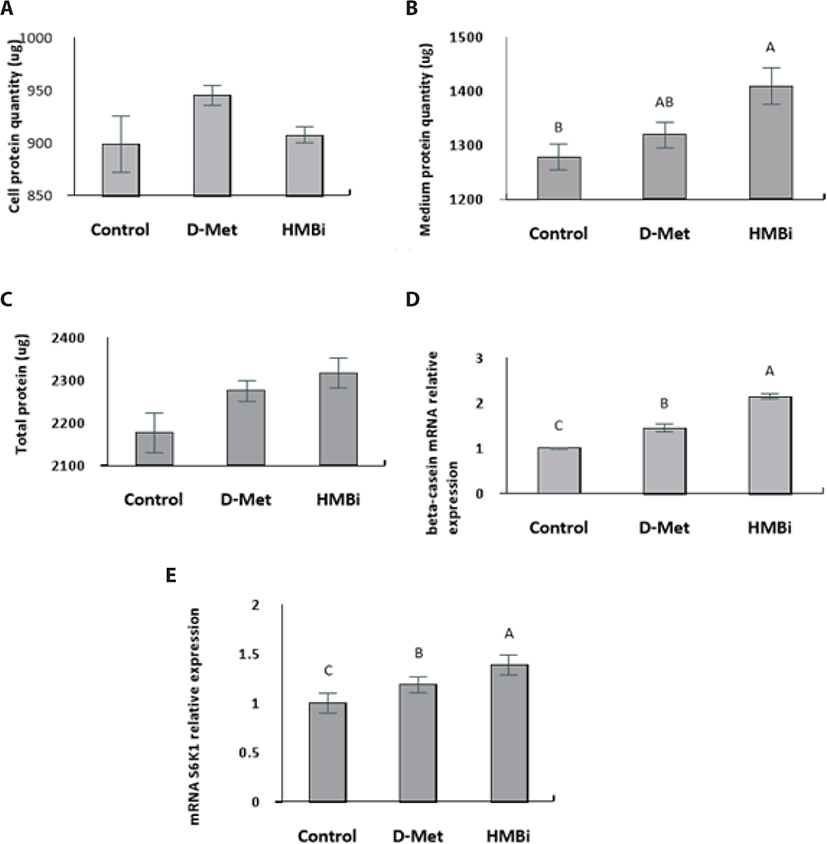
Overall, there were no significant differences in total protein (cell and medium) among the treatments (Fig. 4C; p > 0.05). Following supplementation with D -Met and the Met precursor HMBi at optimal dosage and time, HMBi has the highest beta-casein expression (Fig. 4D; p > 0.05). The addition of HMBi showed the highest gene expression for S6K1, a downstream target of mTOR pathway (Fig. 4E; p > 0.05) [2,7,8].
For the proteomics analysis, the upregulated and downregulated proteins in the D-Met group were 46 and 68, and 40 and 78 in the HMBi group (Table 6). Expression of proteins linked to protein and energy metabolism was altered after supplementations with D-Met and HMBi. The addition of HMBi upregulated the elongation protein eukaryotic translation initiation factor 3 subunit (EIF3A) but downregulated the ribosomal proteins RPS12 and RPS21. Supplementation with D-Met stimulated phosphoinositide 3 (PI3) kinase, an upstream mTOR activator [19,28,33,34]. The list of all upregulated and downregulated proteins are listed in Tables 8 and 9.
HMBi supplementation stimulated the production of EIF3A protein essential for protein translation initiation [19,28] higher than the D-Met group. EIF3A is vital for stimulating protein synthesis [19,28]. The RPS12 and RPS21 were decreased in the HMBi group but not in the D-Met group. The RPS12 and RPS21 proteins are ribosome protein components found in 40S subunit [35]. Increase in EIF3A and the decrease in RPS12 and RPS21 ribosomal protein components when milk protein synthesis is increasing agreed with previous studies [36,37]. It is said that protein export and transformation of AA going to tRNA is increased during lactation involving EIF3A despite a decrease in protein synthesis machinery like RPS12 and RPS21 in the ribosomes. This results in an overall increase in milk protein synthesis and mammary gland milk protein secretion [36,37]. Another study found a decrease in ribosomal protein expression in spite of the increase in milk protein synthesis during lactation [36]. This finding suggests that the decrease or no increase in the number of ribosomal components despite the increase in beta-casein expression and milk protein secretion is a way in which the mammary gland can focus on mRNA translation of proteins related to milk synthesis rather than ribosomes. This allows mammary epithelial cells to synthesize milk protein at an optimal level [36]. The ribosomes in the mammary gland decrease during lactation to favor beta-casein synthesis [36].
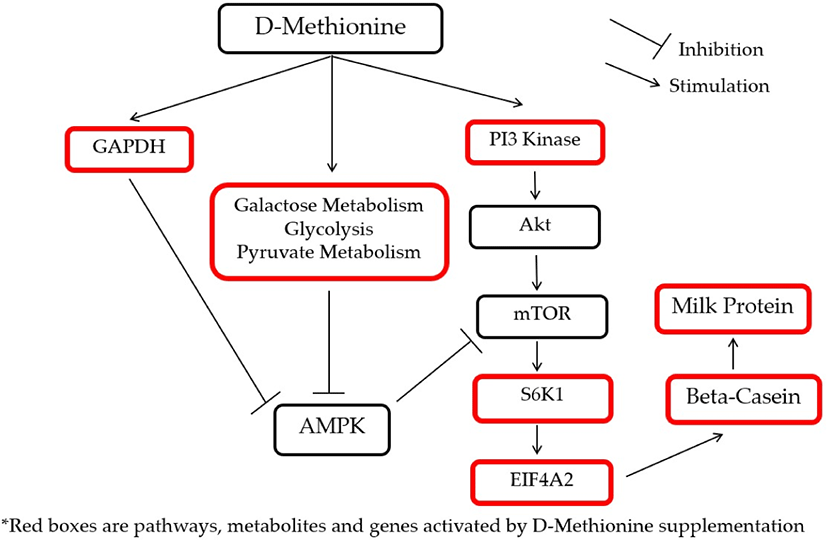
Pathways linked to protein and energy metabolism significantly affected by the supplemental treatments are shown in Table 7. The glycolysis pathway was activated by D-Met and HMBi treatments. The pentose phosphate pathway was activated by HMBi supplementation, and pyruvate metabolism was stimulated by D-Met supplementation. The activation by D-Met supplementation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate metabolism, glycolysis, and galactose metabolism which are energy production pathways will eventually inhibit adenosine monophosphate kinase (AMPK), a known inhibitor of mTOR, and this will result in promoting beta-casein gene expression and increase protein synthesis [38-41]. In addition, the activation of the above-mentioned pathways was expected because the treatment supplementation provides additional energy sources since D-Met and HMBi are known carbon skeleton sources.
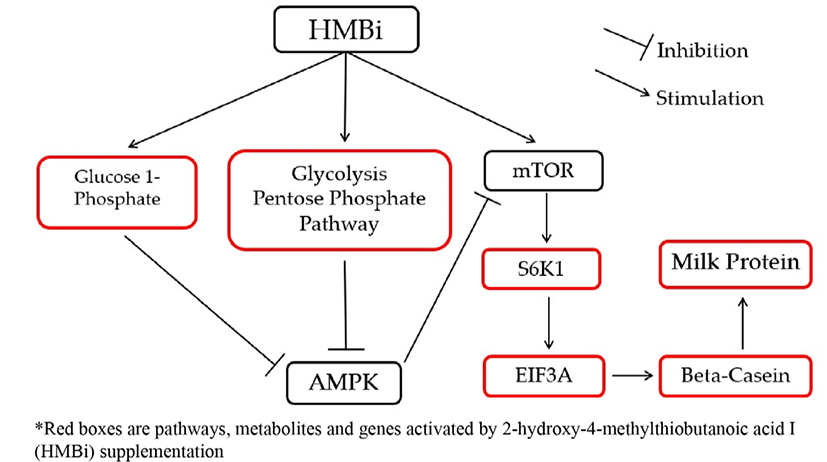
HMBi also stimulated the glycolysis and pentose phosphate pathways, which are involved in metabolic pathways of energy production, and promote increased glucose 1-phosphate which is an energy metabolic intermediate product. These reactions could also influence mTOR stimulation by inhibiting AMPK that will increase beta-casein gene expression and milk protein synthesis [39,40]. The AMPK is a fuel-sensing enzyme that is present in mammalian cells [40]. Previous studies reported that energy intermediates indirectly activate protein synthesis by blocking AMPK, a known direct inhibitor of the mTOR pathway [7,39,40]. An increase in energy status can block the inhibition of AMPK to mTOR [34].
In the analysis of intracellular AA levels inside MAC-T cells, intracellular Met levels in MAC-T cells increased significantly in the D-Met supplemented cells (Fig. 5; p < 0.05). The lack of increase in cell-free Met in HMBi treatment suggests that a small amount of HMBi was converted to Met within the cells. Thus a direct reaction of HMBi with the cell could be possible, as shown in the significantly higher beta-casein and S6K1 gene expression and numerically higher medium protein quantity in HMBi compared to D-Met treatment and control groups.
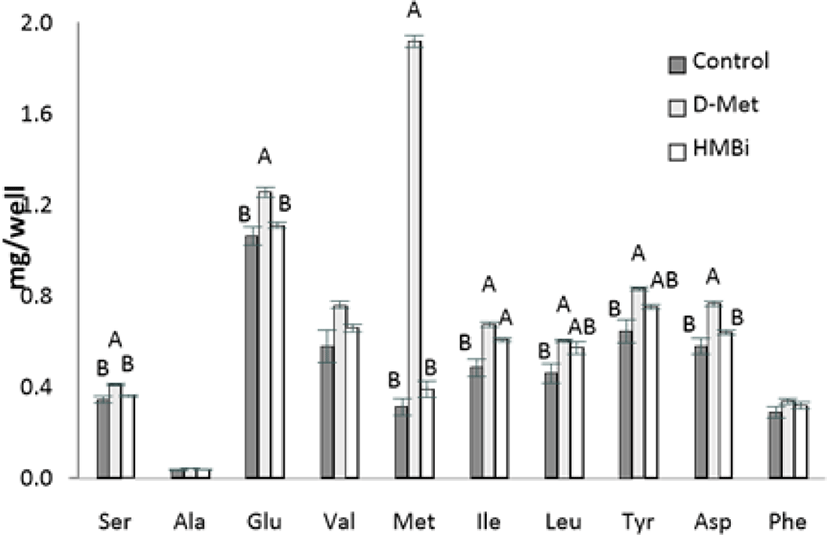
This trend is also seen in serine, glumatic acid, and aspartic acid. There was also a higher concentration of isoleucine found inside the cells in the D-Met and HMBi groups compared to control (p < 0.05). The D-Met group has a higher intracellular concentration of leucine and tyrosine than the control but is not significantly different from the HMBi group (p < 0.05).
Pathway metabolite detection results are listed in Table 10. Most pathways related to the detected metabolites were involved in energy and AA metabolism. Energy pathways such as glycolysis, gluconeogenesis, and the pyruvate metabolism pathway were activated by D-Met supplementation. The detected metabolites affected by supplemental treatments are listed in Table 11. D-Met supplementation increased the production of seven metabolites and decreased UMP. HMBi supplementation increased the production of three metabolites (and decreased that of UMP and N-acetyl-L-glutamate). It also increased the production of the energy metabolite glucose 1-phosphate.
●, Significantly increased pathway metabolites determined by Metabo Analyst v. 3.0 software for Bos taurus, relative to control (p < 0.05) (see Methods for detailed explanation).
To illustrate the interactions of different Met treatments in MAC-T cells, diagrams of the relationships among various components were created (Figs. 6 and 7). Fig. 6 shows the increased relative gene expression of beta-casein, S6K1 and EIF4A2, upregulation of GADPH protein and the stimulation of glycolysis, galactose and pyruvate metabolism that resulted in to increase in milk protein synthesis upon addition of D-Met. Fig. 7 shows that upon the addition of HMBi, there were increases in the relative gene expression of beta-casein, S6K1, and EIF3A, stimulation of glycolysis and pentose phosphate pathways, and increased in the glucose 1-phosphate metabolite.
CONCLUSION
Taken together, these results show that D-Met and HMBi are effective for stimulating milk protein synthesis. D-Met and HMBi supplementation effectively stimulated beta-casein and S6K1 mRNA expression. They are also effective and efficient in activating pathways linked with milk protein synthesis and increasing the expression of proteins and the production of metabolites involved in milk protein synthesis.