
INTRODUCTION
For last decades, microbiota has been focused as a key influencer of the healthy life. Gut microbiota is specified in host’s gastrointestinal environment where provides variable prebiotics of digestible fibers which are able to be used as energy sources by microbiota. Those symbiosis is crucial for the host’s health. There have been many studies that the dysbiosis of gut microbiota is related with the host immune system [1]. Inflammatory bowel disease would be induced by dysbiosis of gut microbiota, Proteobacteria which changes host’s mucus membrane from defensive to pro-inflammatory status [2,3]. Irritable bowel syndrome is also found when gut microbiota composition is disturbed. The balance of gut microbiota composition is also involved in the balance between pro-carcinogenic and anti-carcinogenic colorectal cancer [2,4–6].
The composition and function of the gut microbiota has been reported to be related with obesity in mice and human. In obese mice, the composition of Firmicutes was increased and Bacteroidetes was decreased [7]. Bacteroides species was decreased in obese persons and increased in those person after weight loss by sleeve gastrectomy [8]. There are several studies to support the connection between obesity and gut microbiome. Microbiota produces SCFAs as the end products of fermentation after digesting the non - digestible fibers [9,10] and is used as an energy source of the host [11]. Acetate, which is one of SCFA, transferred to the brain through peripheral circulation that reduced acute food intake [12]. Actually, the concentration of SCFA was higher in overweight subjects [13,14]. In addition, the increase of lipopolysaccharide (LPS) which is a component of microbe’s membrane triggered obesity, inflammation and insulin-resistance when mice were fed high fat diet [15].
Gut microbiota is also involved in dog’s health and diseases. Similar to that observed in other animals the gut microbial composition is related with dog’s metabolism in many processes. In healthy dogs, there are 102–109 CFU/g in the small intestine and 108–1011 CFU/g in the colon of bacteria [16,17]. Dog’s gut microbiota composition was changed by the type of diet like the amount of protein or carbohydrate [18]. High protein and low carbohydrate diets influenced more to microbiome of obese dogs than those of lean dogs [19]. In comparison, the dog microbiota is more similar to the human microbiota than those of pigs or mice according to the metagenomics analysis of microbiome [18]. The dog’s gut microbiome would be more useful to study those in human than pig’s or mice. However, dog’s energy balance does not highly dependent on the microbiota unlike human because they have relatively simpler gastrointestinal tract like other carnivores [20] even though dogs are closer to omnivorous and can utilize a considerable amount of carbohydrates. For dog’s health and well-being, it is necessary to know its own gut microbiota characteristics. Unfortunately, there are limited information about dog’s gut microbiota.
Dogs have been domesticated for a long time and become the major companion animals [21]. Currently, the well-being and longevity of pet dogs has been a main concern of owners and they put a lot of efforts to maintain their dog’s health as one of family members. To keep a healthy life of dogs nutritional management is a crucial. For that, the balanced gut microbiota is important regarding the nutrition absorption and maintaining gastrointestinal health. However, there are still less information of dog’s gut microbiota. In this study we investigated the taxonomic profiles of gut microbiota in dogs. Moreover, we analyzed the difference of taxonomic profiles of gut microbiota based on the body condition scoring system. These finding would contribute to understand the dog gut microbiome diversity and the relationship of gut microbiota with body weight gaining in dogs.
MATERIALS AND METHODS
In total, twenty-four healthy Beagles (24 months of age) were applied. Animal studies were approved by the Institutional Animal Care and Use Committee of the National Institute of Animal Science, Korea (Approval number: NIAS20191665). Dogs were fed with a commercial diet (33% crude protein, 20% crude fat, 3% fiber, 13% crude ash, 1% calcium, 0.8% phosphorous, 12% moisture and 4,100 kcal/kg of ME) based on the metabolic body weight twice a day and water with free assess. Dogs were housed in a single kennel and exercised outdoor twice a day. Dogs have been maintained routinely and there was no additional treatment or supplement diet for this study. All dogs were monitored every day and examined by veterinarians if needed.
Fresh feaces were collected and directly frozen in liquid nitrogen. After delivered to the laboratory feaces were stored at −80°C until used. Genomic DNA (gDNA) was extracted from frozen feaces by using NucleoSpin®DNA Stool (Macherey-Nagel, Germany). Briefly, 180–220 mg of a fecal sample were used and lysed using MN Bead Tube Holder and the gDNAs were harvested by NucleoSpin®DNA Stool Column centrifuging at 13,000 ×g for 1 min. At the final step gDNA was eluted in the elution buffer after centrifuging at 13,000 ×g for 1 min. gDNAs were stored at −20°C.
Sequences of 16s rDNA at V3 and V4 hypervariable regions were targeted to amplify according to the previous study [22]. To target the V3 and V4 regions the full length primer sequences were used as below: forward primer 5’–TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG–3’ and reverse primer 5’–GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC–3’. Illumina adapter overhang nucleotide sequences were added at the gene-specific sequences. Locus specific sequences were as forward overhang 5’–TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG–3’ and reverse overhang 5’–GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG–3’. To purify PCR products KAPA HiFi HotStart ReadyMix (KAPA Biosystems, USA) and Agencourt AMPure Xpsystem (Beckman Coulter Genomics, USA) were used. The libraries are sequenced on the Illumina MiSeq instrument (2 × 300 paired-end sequencing).
After sequencing the original paired-end reads were trimmed by removing the adaptor sequences with CutAdaptV1.11. The merged reads were generated with the first processed paired-end reads using FLASH v1.2.11. For the quality control low quality merged reads were removed if they contain two or more unclear nucleotides, have less than 20 average quality scores and have shorter than 300 bp length after trimming low quality bases. At the final stage, the chimeric reads were removed using ChimeraSlayer r20110519. The number of operational taxonomic units (OTUs) were calculated by using the pre-processed reads from each sample and determined by clustering the sequences based on 97% of the sequence identity using QIIME software (v.1.8.0).
Taxonomic abundance was calculated with cd-hit v.4.6 by clustering consensus sequences which have over 99% of identity and 80% of coverage. Using megablast algorithm the consensus sequences were aligned to the nucleotide database of NCBI. Taxonomic profiling was performed for assembled genome based on NCBI taxonomy information and KronaTools v.2.7.
The alpha diversity for each sample was measured by OTUs using Shannon index, and the beta diversity was measured by the difference in organism composition measured according to Bray-Curtis distance. The differences among microbiota communities based on the BCS were evaluated by One-way ANOVA. Statistical significance was determined if p was less than 0.05.
RESULTS AND DISCUSSION
Overweight and obesity are related with the development of numerous diseases. Nutritional management in dogs have been a critical issue to maintain their healthy life and wellbeing by monitoring the optimal body weight. Currently it has been reported that obesity can affect the gut microbial composition in mice [7]. Microbial community was also differently existed depending on BMI scores in human gut [23]. Obese people had fewer Bacteroidetes and more Firmicutes [24]. For that reason, Turnbaught et al. suggested that obese microbiome increased the capacity of the energy harvest from foods that would cause the accumulation of excessive energy in body [13]. This finding was supported by another study that the total amount of SCFA (the end product of fermentation by gut microbiota) was higher in overweight people [14]. Similar to human, gut microbiota was different compared to those in lean dogs and obese dogs, and SCFA was lower in dogs with faster weight loss. However, there is little information available about gut microbiome related with body weight gaining in dogs. Therefore, we investigated the difference of gut microbial compositions based on the body condition scores using metagenomic analysis in healthy dogs.
Metagenomic analysis is a powerful method to provide insights into the whole microbiota community residing a certain environment. The survival and function of microbes were involved with host and the complexed microbial community rather than individual microbe itself. Here, to study the composition of microbial community whole microbiome in dog’s gut was analyzed by using 16s rRNA sequences. The twenty-four healthy Beagles were examined their body condition scores (BCS) with which they are grouped into BCS 3 (lean), 4–5 (optimal) and 6 (overweight). BCS is an indirect method to estimate the body fat accumulation. The number of reads after the preprocessing of sequencing data was shown in Fig. 1A. The operational taxonomic units were higher in BCS 3 (lean) compared to those in BCS 4–6 (Table 1). The alpha diversity was decreased when dogs were overweight (Table 1). It implied that the diversity of microbiome in dogs would have an opposite relationship with BCS.
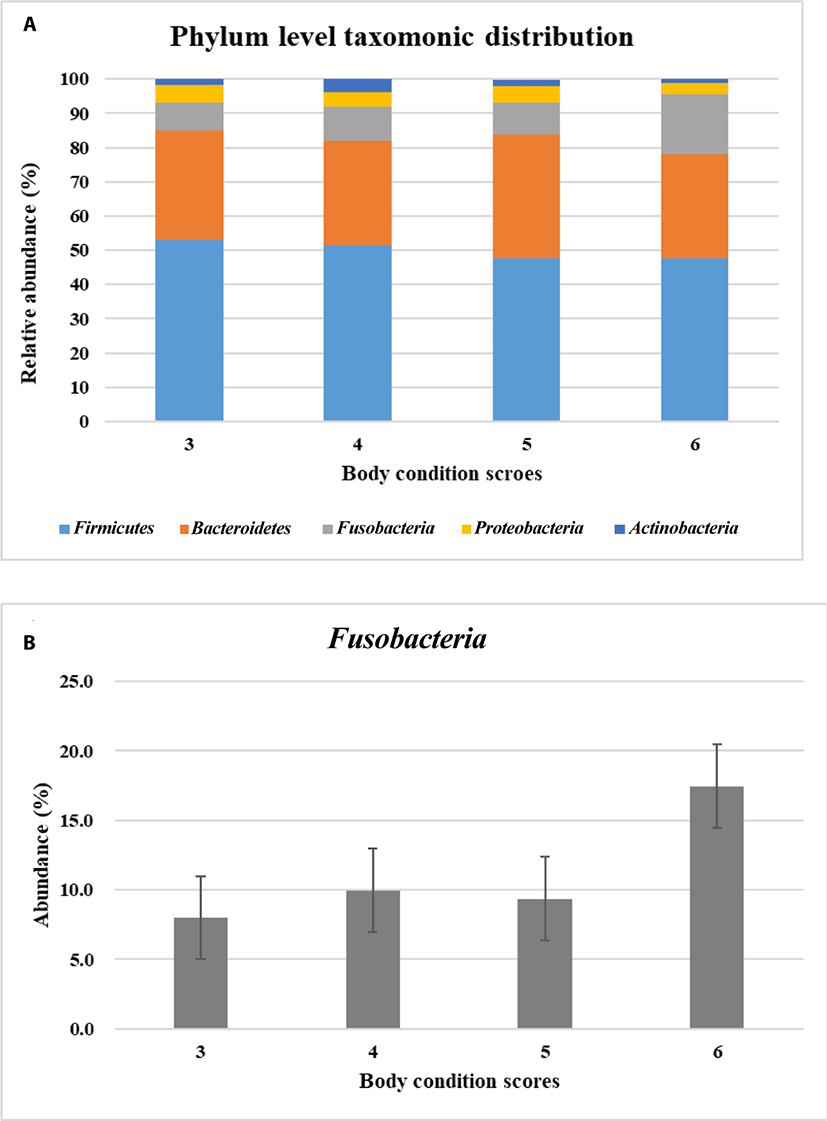
The predominant microbial phyla for maintaining gastrointestinal health includes Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria and Actinobacteria in dog’s gut [25–26]. In taxonomic profiles of metagenomics samples, Firmicutes, Bacteroidetes, Fusobacteria, Proteobacteria, and Actinobacteria were predominant as previous reports in this study. Next, these five predominant microbiota were investigated how they were distributed based on the BCS. Phylum level taxonomic distribution of the predominant microbiota was different in dogs according to BCS (Fig. 1A). Among them Fusobacteria had tendency to be more abundant when dogs were overweight in BCS 6 (not statistically significant) (Fig. 1B). Actually, Firmicutes was well known as the main obese microbe in human and mice rather than Fusobacteria [7,24]. Fusobacteria is anaerobic, gram negative, nonsporulating, nonmotile and long spindle shaped rods [27]. It is known to have little fermentable ability, be infectious and induce inflammation [27]. Fusobacteria can be found in the oral cavity, gastrointestinal tract, and female genital tract. In human Fusobacteria has been known to be related with diseases like colorectal carcinoma, acute otitis or sarcoidosis. However, there is little information available about the function of Fusobacteria in dogs.
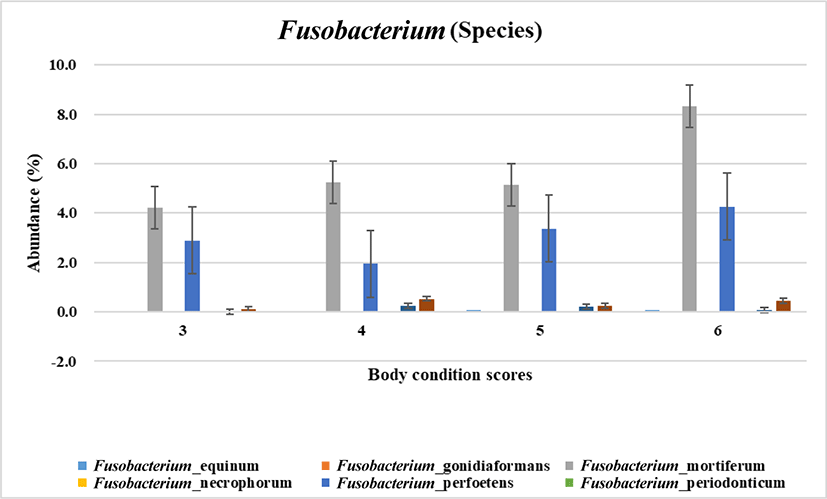
To narrow down the involvement of fusobacteri at species level regarding the body weight gain microbial species was analyzed. As the result shown in Fig. 2, Fusobacterium mortiferum and Fusobacterium perfoetens were highly related with increased body condition scoring. According to the statistical analysis F. perfoetens resided differently in dogs with the optimal and overweight body conditions (p < 0.05). Unlike other Fusobacterium species which are weaker fermenters F. mortiferum is able to use a variety of sugars as energy sources [28]. F. nucleatum is the most common Fusobacterium specices isolated from humans and known to be related with infections and colorectal carcinoma [6]. Unlike other Fusobacterium. F. perfoetens has the coccoid morphology rather than the long spindle rod shape of the typical morphology of Fusobacterium [29]. In phylogram showing genetic relationship, F. perfoetens formed a separated branch from the clusters of other Fusobacterium species [29]. In the view of the facts previously reported F. perfoetens (including F. mortiferum in a certain aspect like the energy utilization in gut) would have non - typical and different function in gut microenvironment unlike human. Therefore, it is possible that the gut microbiome in dogs would be relatively unique although it shares a similarity with human. However, an additional quantificational experiment is needed for the further verification of the relationship between Fusobacteria and obese which we suggested in this study.
Dogs have been considered as one of family members. Dog’s well-being has also become an important issue in the human society. Dog’s owners are well aware the importance of balanced nutritional management and give an effort to provide a good quality and quantity of diets to their dogs. However, the comprehensive information of dog’s gut microbiota that affects on the overall dog’s health is still in need. In this study, we investigated the difference of dog’s gut microbiota according to the body condition scoring system which reflects the level of the body fat accumulation condition. We found dog’s gut habits microbiota differently based on the degree of body weight gain. This finding would contribute to improve and develop the nutritional knowledge of gut microbiome for dog’s healthier life.