













INTRODUCTION
Bovine viral diarrhea (BVD) is an economically significant disease in cattle and is found in most countries worldwide. Infection and death associated with BVD lead to significantly reduced reproductive performance and increased premature culling. These clinical signs are especially pronounced when one or more BVD carriers are present in a herd. Animals that develop acute diarrhea and fever may die or have long, costly recovery periods with decreased production and growth performance. Over the past 10 years, BVD has occurred frequently, causing economic losses to farmers in the domestic livestock industry [1]. The major pathogens causing calf diarrhea in these reports were viruses (bovine coronavirus [BCV], bovine rotavirus group A [BRV], and bovine viral diarrhea virus [BVDV]), bacteria (Escherichia coli K99 and Salmonella spp.), and protozoa (Cryptosporidium parvum and Eimeria spp.). Some of these agents are detected not only in diarrheic calves but also in normal calves [2,3]. BVDV is predominant in most parts of the world, with a high prevalence, persistence, and clinical consequences [4]. BVDV is an enveloped, positive-sense, linear, single-stranded RNA virus (12.5 kb) of the genus Pestivirus in the family Flaviviridae. BVDV can be divided into two types (BVDV1 and BVDV2) based on sequence similarity of the 5’ untranslated region (UTR) in the viral genome [5]. BVDV transmission occurs both horizontally and vertically, with persistently and transiently infected animals excreting the virus. The virus is transmitted via direct contact, bodily secretions, and contaminated fomites, and can persist in the environment for more than two weeks. Persistently infected animals are the most important source of the virus, continuously excreting a viral load 1000 times higher than that shed by acutely infected animals [6]. Laboratory testing for identifying BVDV has typically been performed with methods such as enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR). However, these are costly and require precise instruments, making it imperative to establish a fast, accurate, and efficient method for detecting BVDV [7–9]. Quick and accurate point-of-care (POC) testing for BVDV would greatly enhance diagnostic capacity, leading to improved quarantine and disease prevalence control. There are several POC RNA detection technologies that do not require special instruments, such as reverse transcription recombinase polymerase amplification (RT-RPA) [10] and reverse transcription-loop-mediated isothermal amplification (RT-LAMP) [11]. RT-RPA and RT-LAMP are highly sensitive methods for detecting viral RNA; however, they are prone to non-specific amplification under isothermal conditions. This can lead to false positive results when used for viral RNA detection [12]. This problem worsens when non-sequence-specific probes, such as pH-sensitive dyes, are used. The use of sequence-specific probes, such as hybridization-based fluorescent oligonucleotide probes, can improve detection sensitivity [13]. Diagnostic methods have been developed based on clustered regularly interspaced short palindromic repeats (CRISPR), which utilize the collateral cleavage activity of bystander nucleic acid probes by RNA-guided CRISPR-associated 12/13 (Cas12/13) nucleases [14]. CRISPR diagnostic methods are both highly sensitive (at the attomolar level) and specific (down to the single-nucleotide level) [15]. The readout of Cas-mediated nucleic acid probe cleavage can be detected using fluorescence or the lateral-flow strip method. The latter is advantageous because the strips are portable, and the results can be easily read with the naked eye [16]. Cas13a, previously known as a single-effector RNA-guided ribonuclease (RNase), can detect the presence of an RNA target using CRISPR RNAs (crRNAs), providing a platform for specific RNA sensing [17]. Several studies have reported that Cas13a-based molecular detection systems can detect both Zika and Dengue viruses [15]. Recently, a nucleic acid detection method based on CRISPR-Cas13a was developed as a new system for the early diagnosis of BVDV [9]. This study aimed to establish a nucleic acid detection method based on the lateral flow strip method for early POC detection of BVDV-1b using CRISPR-Cas13a. To improve the accuracy and performance of POC diagnosis in testing settings with limited resources, we designed a crRNA in the 5’UTR conserved region and a sequence-specific primer to provide detection.
MATERIALS AND METHODS
MDBK cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin at 37°C with 5% CO2. MDBK cells were obtained from Korean Cell Line Bank, and BVDV-1b cells were obtained from the Korea Veterinary Culture Collection (KVCC). MDBK cells at 80%–90% confluency were washed twice with phosphate-buffered saline (PBS) and then infected with BVDV-1b diluted to a concentration of 102–103 using DMEM. The cells were then incubated at 37°C with 5% CO2 for 60–90 minutes to allow the virus to adsorb onto them. Infected MDBK cells were cultured for more than 48 h in DMEM supplemented with 3% FBS. The cells were observed daily for cytopathic effects (CPE). The cells were lysed by three repeated freeze-thaw cycles, and the lysate was centrifuged for 10 minutes at 494×g in a 15 mL tube. The supernatant was aspirated and stored at -70°C until further use. The viral RNA of BVDV-1b was extracted using the Viral Gene-spin Viral DNA/RNA Extraction Kit (iNtRON Biotechnology, Seongnam, Korea), following the manufacturer’s instructions.
The extracted viral RNA of the BVDV-1b was reverse transcribed into complementary DNA (cDNA) using the AccuPower® RT Master Mix (Bioneer, Daejeon, Korea), following the manufacturer’s instructions. The template RNA, forward primer (5’-GCCATGCCCTTAGTAGGACT-3’), reverse primer (5’-T7 promoter sequence-CGAACCACTGACGA CTACCC-3’), RNase inhibitor, and ddH2O were combined in a 1.5 mL tube. The mixture was incubated at 70°C for 5 min and then placed on ice. AccuPower® RT Master Mix and DNA polymerase (Bioneer) was added to the PCR tube to achieve a final volume of 50 µL. The RT-PCR reaction was performed by incubating the mixture at 42°C for 60 minutes, followed by 5 minutes at 95°C and then 35 cycles of DNA amplification, which consisted of 30 seconds at 95°C, 30 seconds at 54°C–67°C, and 1 min at 72°C. A final extension step of 5 minutes at 72°C was included. The cDNA was stored at −20°C until use.
The 5’UTR region of the BVDV FASTA sequence (NC_001461.1) was obtained from National Center for Biotechnology Information (NCBI). Forward and reverse primers were designed using the NCBI Primer-BLAST. For detection reactions involving Cas13, a T7 promoter sequence must be added to the 5′ end of the forward or reverse primer to enable T7 transcription. The gRNA sequence was designed using the Cas13 online platform (https://cas13design.nygenome.org). The 5′ end of the gRNA sequence is then combined with a direct repeat (DR) sequence to form a complete crRNA [17]. The primers and crRNAs were designed as shown in Table 1.
The pC013-Twinstrep-SUMO-huLwCas13a plasmid (#90097, Addgene, Watertown, MA, USA) was transformed into Rosetta™2 (DE3) pLysS Single Competent Cells (NovagenTM Sigma-Aldrich, St. Louis, MO, USA). The transformed cells were plated on an Luria-Berani (LB) agar plate and incubated for 24 hours at 37°C. The following day, a single colony was isolated and cultured in 25 mL of Terrific Broth (TB) medium with 12.5 µL of ampicillin at 300 rpm for over 9 hours at 37°C until an optical density (OD)600 of 0.4–0.6 was reached. The culture was then cooled at 4°C for 30 minutes. After adding 62.5 µL of 0.2 M Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich), the cells were incubated in shaking incubators at 300 rpm for 16 hours at 21°C. After 16 hours of cultivation, the cells were centrifuged at 5,000 rpm for 15 minutes at 4°C. The cell pellet was then stored at −80°C until it was used.
The cell pellet was resuspended in 20 mL of supplemented lysis buffer (composed of 50 mL of lysis buffer, 1 tablet of cOmpleteTM Ultra EDTA, 0.05 g of lysozyme, and 0.5 µL of benzonase) by stirring the mixture on ice for 30 min. The resuspension was sonicated using a Vibra cell (Sonics, Newtown, CT, USA) for 10 min with a 1-second on and 2-second off cycle at a 30% pulse amplitude. The lysate was cleared by centrifugation at 10,174×g for 30 minutes at 4°C, after which the supernatant was transferred to a new 50 mL conical tube. The supernatant was added to 1.25 mL of Strep-Tactin Superflow Plus resin (Qiagen, Hilden, Germany), and the mixture was gently shaken at 4°C for 2 hours to bind the Twinstrep-SUMO-huLwCas13a protein to the resin. The glass Econo-column® (BIO-Rad, Hercules, CA, USA) was prepared by washing the column with cold lysis buffer (4 mL of Tris-HCl [1M, pH8.0], 20 mL of NaCl [5M], and 0.2 mL of DTT [1M], adjusted to a final volume of 200 mL with ddH2O) on ice. The solution was poured and allowed to flow through the column. The column with the binding resin was washed by adding 18.75 mL of cold lysis buffer. Then, 3.75 mL of SUMO protease cleavage solution (3 μL of SUMO-protease, 7.5 μL of IGEPAL® CA-630, and 5 mL of lysis buffer) was added to the column and it was covered. The mixture was gently shaken at 4°C for 16 h. The SUMO cleavage sample was collected in a new 50 mL conical tube, and 1.5 mL of glycerol was added. The LwCas13a protein was stored at −20°C.
For each Cas13-based detection reaction, a pre-cooled 1.5 mL microcentrifuge tube was used, and the following components were added:3.5 µL DEPC-treated water, 1.2 µL Tris-HCl (1M stock, pH 8), 6 µL MgCl2 (20 mM stock), 0.8 µL NaCl (1M stock), 2 µL BVDV-1b cDNA, 2 µL LwCas13a, 1 µL Murine RNase inhibitor (40 U/µL stock; New England Biolabs, Ipswich, Massachusetts, USA), 0.6 µL T7 RNA polymerase (50 U/µL stock, Bioneer), each 0.5 µL rNTPs (ATP,GTP,CTP,UTP) (10 mM stock, Bioneer), and 0.4 µL RNA reporter (10 µM stock, Bioneer). And then, a 0.2 mL reaction tube containing 0.5 µL crRNA (1 µM stock) was added to the mixture to achieve a final volume of 20 µL. A negative control (NC) was prepared without crRNA. The reaction tubes were incubated at 37°C for 6 hours to allow reaction of T7 transcription and collateral cleavage at the same time. To analyze the Cas13-based reaction using a lateral-flow strip, 80 µL of HybriDetect assay buffer was added to each reaction tube containing Cas13 enzyme and mixed thoroughly. And then, 25 µL of the detection solution was dropped onto the sample spot of a HybriDetect lateral-flow strip (Milenia Biotec, Gießen, Germany) allowing the reaction to flow into the strips. We used gold nanoparticles (GNPs) provided by HybriDetect lateral-flow strip (Milenia Biotec).
RESULTS
The RNA genome of BVDV-1b is approximately 12.5 kb in length containing a single open reading frame (ORF) flanked by a 5’ and 3’ UTR [18–20]. As shown in Fig. 1A, five structural proteins of BVDV (nucleocapsid; C/BVDV-encapsulated glycoprotein; Erns, E1, E2/ ion channel-related protein; P7) were present in the N-terminal region. Six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are located in the C-terminal region. The BVDV protein sequence contained NH2-Npro-C-Erns-E1-E2-P7-NS2-NS3-NS4ANS4B-NS5A-NS5B-COOH [21–23]. BVDV presentation (processing) begins with structural/non-structural protein degradation by Npro (autoprotease) in the N-terminal region [21]. The 5’ UTR of BVDV-1b was selected as the molecular detection region due to sequence conservation. BVDV was classified into BVDV-1 and BVDV-2 based on the nucleotide sequence of the respective 5’ UTR region [5]. As a result of observing MDBK cells infected with BVDV-1b after 48 hours, the biotype of the virus was non-cytopathic.
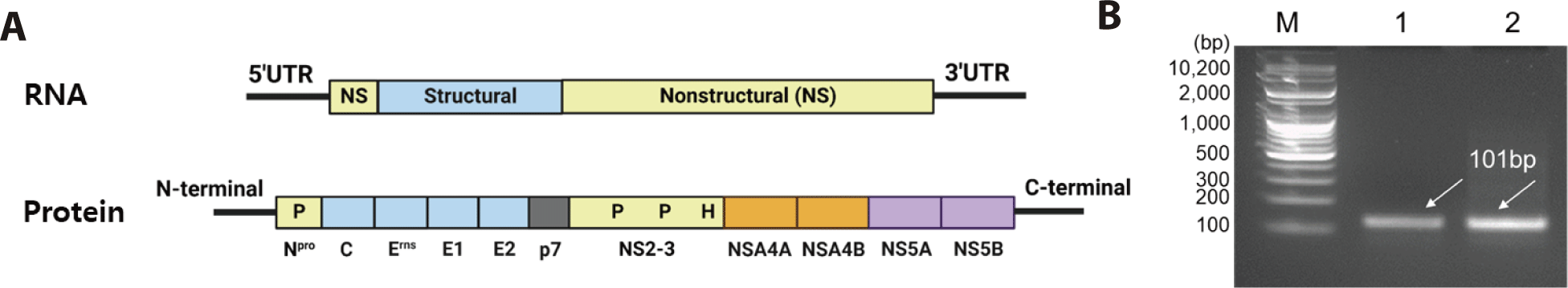
The cDNA was synthesized by RT-PCR targeting the 5’ UTR region of BVDV-1b, resulting in a 101 bp product (Fig. 1B). Furthermore, Fig. 2 shows the workflow for detecting the cDNA using the CRISPR-Cas13 system. The cDNA must be transcribed to bind with the crRNA. As shown in Fig. 3A, the crRNAs did not overlap with the primers. The crRNAs were designed similarly. The sequences of the primers and crRNA were designed relative to the target cDNA and transcribed RNA. A T7 promoter added to the 5′ end of the primer, and the crRNA sequence was the reverse complement of the target site in the transcribed RNA. The DR of the LwCas13a crRNA was located at the 5’ end of the spacer sequence, forming a complex with the Cas13 enzyme (Fig. 3B).
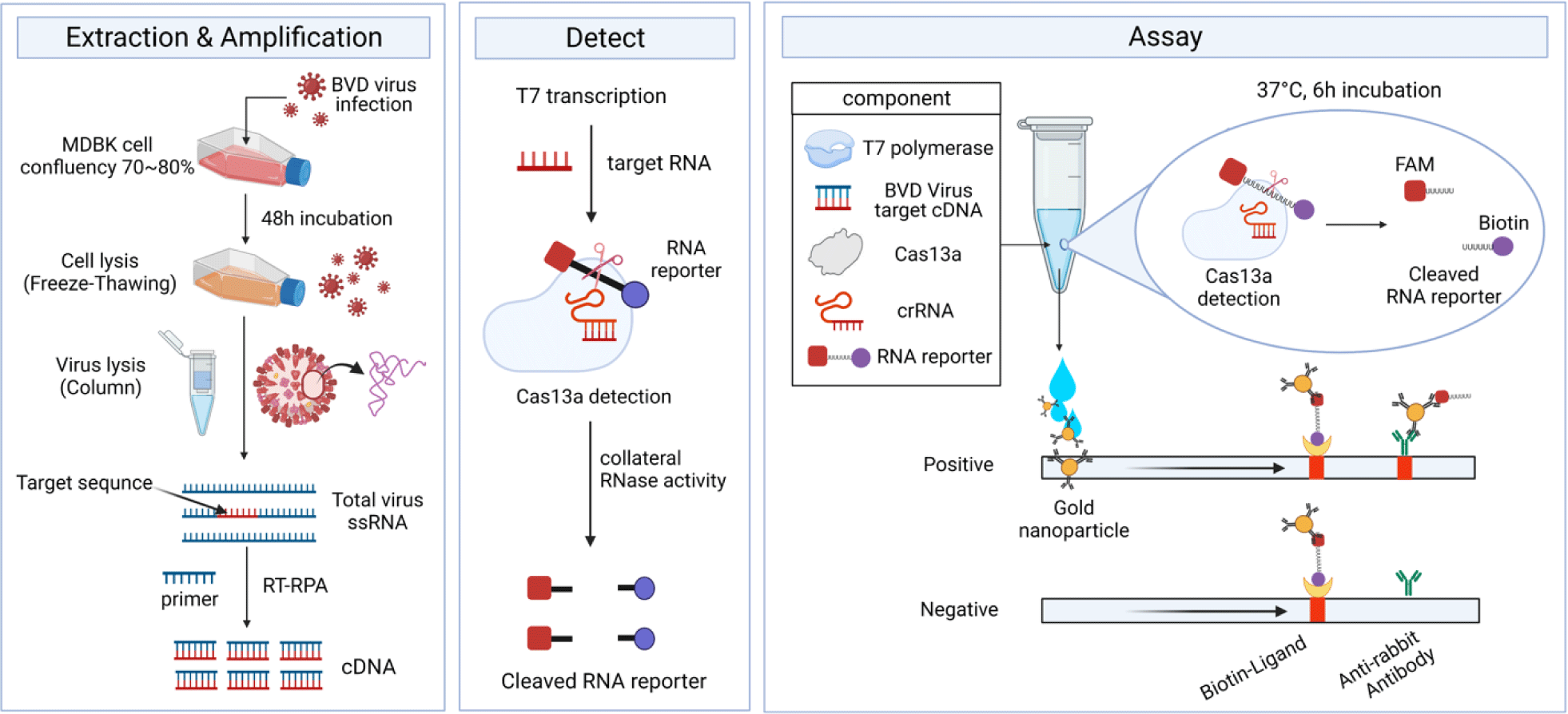
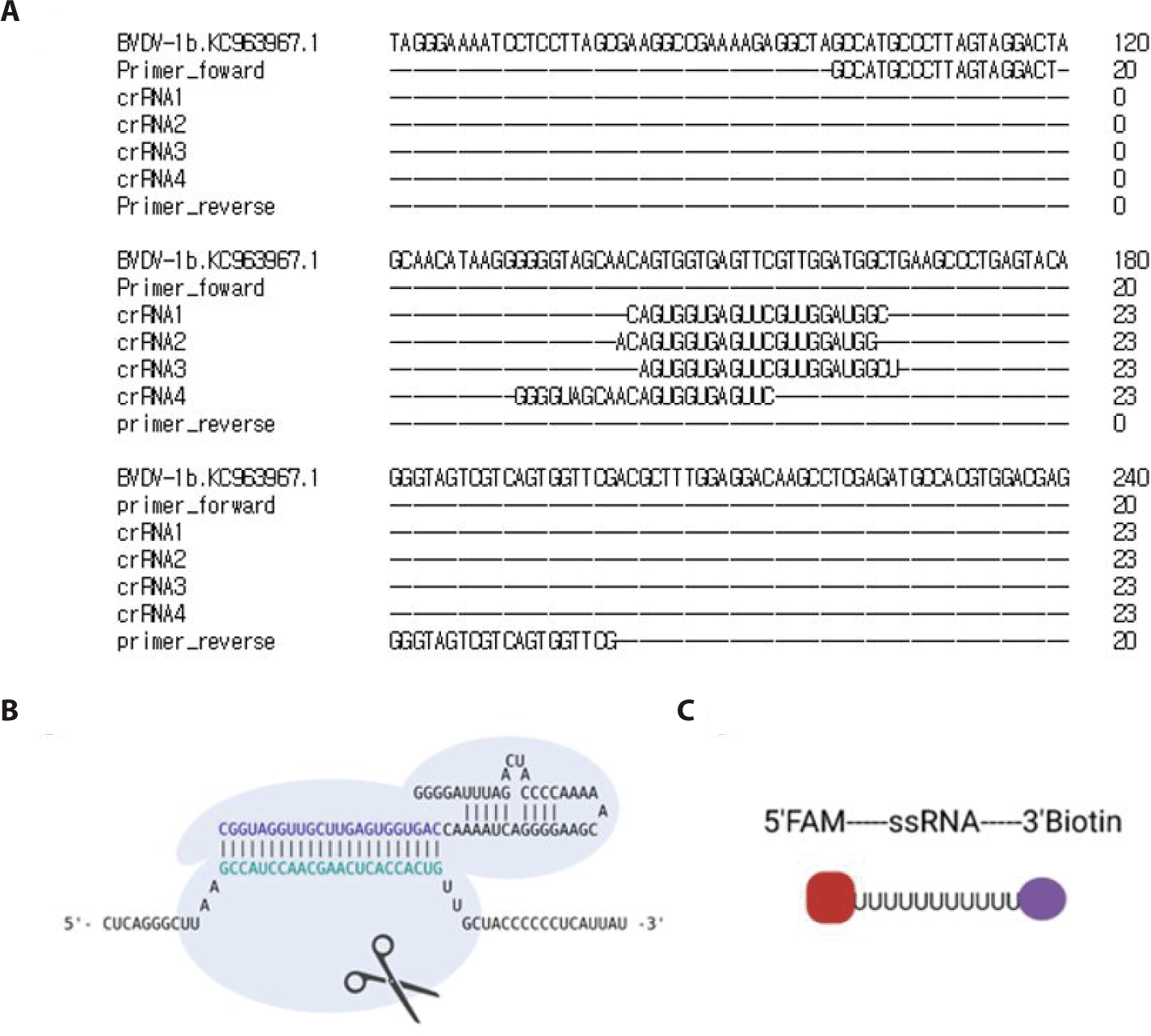
The HybriDetect lateral flow strip (Milenia Biotec) used for the detection assays consisted of a biotin ligand on the control line and an anti-rabbit antibody on the test line. GNPs were coated on the sample spot of the strip, which produced a color. The GNP can bind to fluorescein amidite (FAM) or the Anti-rabbit antibodies on the test line. As a colorimetric-based lateral-flow detection assay (LFDA) probe, an RNA reporter was labeled with FAM at the 5′ end of the poly U sequence and Biotin at the 3′ end (Fig. 3C). Thus, the biotin of the RNA reporter bound to the biotin ligand on the control line of the strip, and the FAM of the RNA reporter bound to the GNP (Fig. 4A).
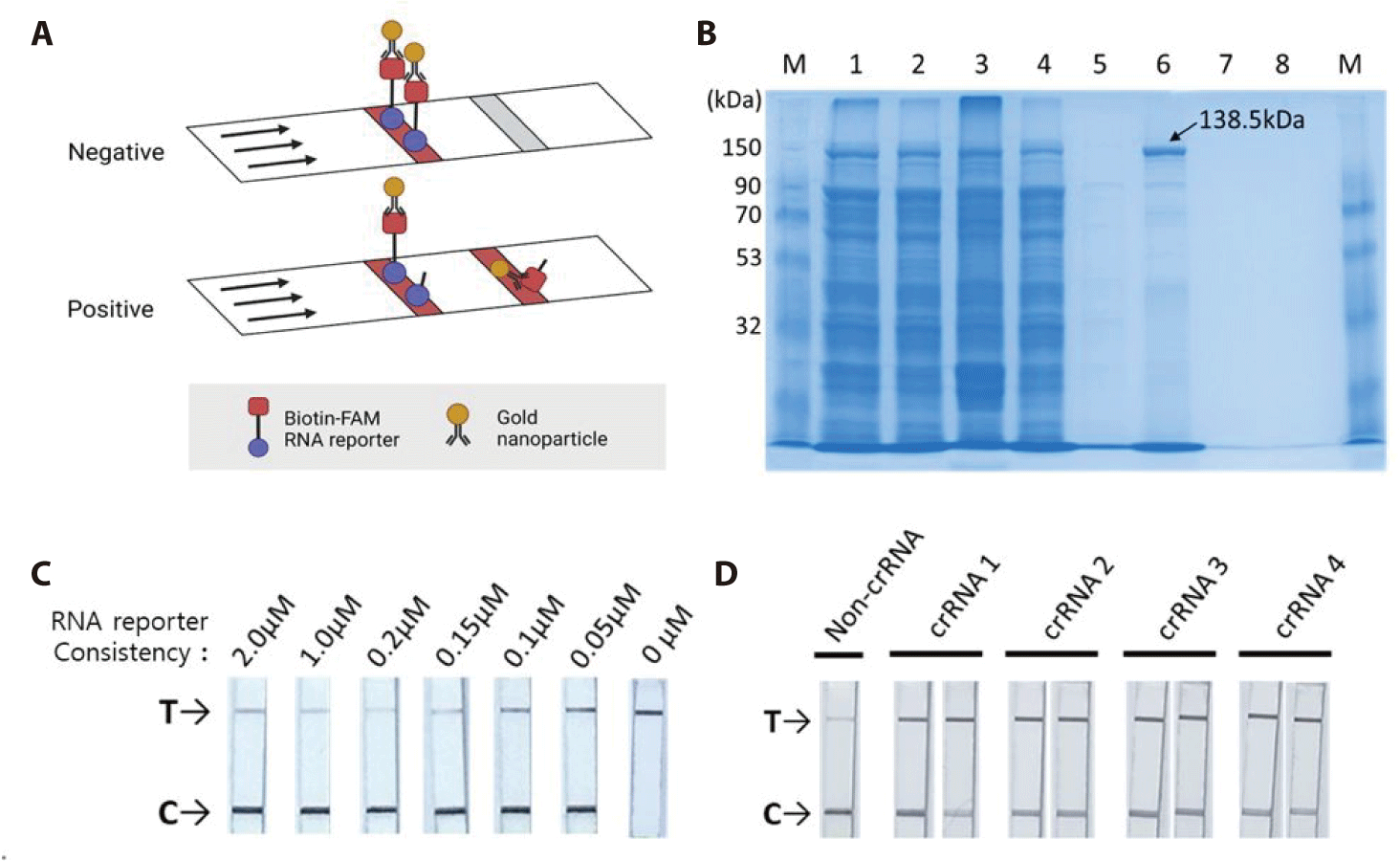
The pC013-Twinstrep-SUMO-huLwCas13a plasmid (#90097, Addgene) was digested into fragments using Xho I. RosettaTM (DE3) was transformed with the plasmid, and single colonies were obtained for the analysis of LwCas13a protein expression. The purified LwCas13a protein showed a single band on SDS-PAGE with a molecular weight of 138.5 kDa (Fig. 4B), indicating successful purification, and was expressed primarily in the supernatant [9].
To confirm the collateral cleavage activity of Cas13, we performed a colorimetric LFDA. The results of the colorimetric-based LFDA using the CRISPR-Cas13 system for detecting BVDV-1b are shown in Figs. 4C and 4D.
First, we tested the RNA reporter at a concentration of 0 µM, and only a band appeared on the test (T) line (Fig. 4C). The GNP did not bind to the control (C) line without the RNA reporter. The Biotin-FAM RNA reporter specifically binds to GNP. To avoid false positive and high dose hook effect (a state of antigen excess relative to the antibody proves, resulting in falsely lowered values), we tested the RNA reporter at concentrations of 2 µM, 1 µM, 0.2 µM, 0.15 µM, 0.1 µM, and 0.05 µM using a LFDA with a paper strip. All lateral flows with paper strips contained test (T) and control (C) bands. We found that a concentration of 0.2 µM resulted in a true-negative readout.
Secondly, we verified that all four crRNAs could effectively detect BVDV-1b. As LwCas13a showed RNase collateral activity, we designed a Cas13a-responsive RNA reporter that remained susceptible to ssRNA-mediated cleavage (Fig. 3C). The RNA reporter was labeled with biotin and FAM, allowing it to be captured on a lateral flow strip with a biotin ligand and detected using anti-FAM antibodies conjugated to gold nanoparticles (biotin-ligand-FAM-GNP). In the true-negative control (no crRNA), all the intact biotin-FAM-GNPs bound to the biotin ligand were captured in the control (C) line. In a positive sample, the reporter is cut, releasing the FAM-containing fragment to be captured by a second line of antibodies resulting in a “test band.” Specific detection of the RNA target cleaves the biotin-FAM reporter, allowing the production of two colored bands in the test (T) line (where anti-rabbit IgG binds to excess anti-FAM-GNPs). We tested the detection of BVDV-1b using a colorimetric LFDA with the CRISPR-Cas13 system (Fig. 4D). True-positive samples produced two colored bands in the control (C) and test (T) bands, which were distinguishable from the true-negative controls (main band at C). Analysis of BVDV-1b showed the specificity of the BVDV-1b gene detection.
DISCUSSION
BVDV-1b is an economically significant viral disease in the national cattle industry. BVDV has spread quickly throughout farms, making prevention and control of the virus challenging [24,25]. Therefore, early POC testing is essential to prevent and control the spread of BVDV-1b. The nucleic acid detection method has the advantage of high sensitivity and is more sensitive and specific than the BVDV-1b antibody detection method. As a result, RT-PCR of viral RNA has been widely used in various countries to detect BVDV-1b. RT-PCR is regarded as the essential method for the diagnosis of BVDV-1b and has become the gold standard for diagnosis in the Republic of Korea. Therefore, Diagnosing BVDV-1b using RT-PCR has a limited ability to prevent and control the spread of BVDV-1b throughout the farm, it requires expensive equipment, such as a thermal cycler, and skilled personnel to conduct the experiments. POC testing has become an excellent supplement to the gold standard for diagnosis using RT-PCR in standard laboratories. Recently, a nucleic acid detection method based on CRISPR-Cas13a was developed as a new system for the early diagnosis of BVDV [9]. According to several studies, nucleic acid detection based on the CRISPR-Cas13 system is a novel strategy for developing detection of RNA viruses for POC testing [14–16,26]. Patchsung et al. [26] validated the SHERLOCK method on 154 clinical COVID-19 samples and found it to be 100% specific, 96% sensitive with a fluorescence readout, and 88% sensitive with a lateral-flow readout. This method was able to detect SARS-CoV-2 in RNA extracts from nasopharyngeal and throat swab samples, including sputum samples, without any cross-reactivity to other common human coronaviruses, and was able to detect the virus in asymptomatic cases [26]. Myhrvold et al. [16] have validated the ability of SHERLOCK to detect Dengue virus (DENV) and Zika virus (ZIKV) infections. All RT-PCR-positive ZIKV and DENV RNA samples were confirmed to be positive for ZIKV and DENV after 1hour of detection [16].
BVDV strain was classified based on a variety of genetic variance in the 5′ UTR. Among the BVDV strains, BVDV-1b occurs frequently in the Republic of Korea. This information was obtained from the Animal and Plant Quarantine Agency. We found that all crRNAs detected were BVDV-1b. Hence, the nucleic acid detection method using the CRISPR-Cas13 system could be useful for POC testing of BVDV-1b as a novel strategy in the Republic of Korea.
To meet the World Health Organization’s “ASSURED” criteria for diagnostic tests – affordable, sensitive, specific, user-friendly, rapid, equipment-free, and deliverable, several challenges must be addressed. Ideal POC testing products must be characterized by miniaturization, automation, visualization of results, and rapid, high-precision, and high-throughput detection. Several problems are associated with POC testing using the CRISPR-Cas13 system. First, the SHERLOCK reagents, which are detection reagents from BVDV-1b, must be freeze-dried not only for cold chain transportation but also for long-term storage or easy reconstitution on paper strips for field applications. Another problem that combines LwCas13a collateral cleavage activity with lateral flow readings is disruption of the FAM-biotin reporter gene. There is increasing evidence that combining Csm6 with LwCas13a detection can improve the test stability and reduce the possibility of false-positive readings [16].
As shown in Figs. 4C and 4D, a faint line was observed on the lateral flow paper strip. Kim et al. [27] observed incomplete removal of the test line from the colorimetric assay results of the negative control, which could lead to false-positive results. When using the traditional colloidal gold test strip for immunological testing, false-positive results are commonly observed, and these results may have serious consequences that affect clinical diagnosis and subsequent control measures [27]. To reduce the false-positive rate of the ERASE assay, Li et al. [28] adjusted its interpretation mode and considered the disappearance of the T-band as the positive threshold. Casati et al. [29] empirically determined that positive samples exhibit band intensity ratios greater than 0.2 compared to negative samples.
CRISPR-based nucleic acid detection, multi-silicon microfluidic chips, and multi-isothermal PCR-based molecular diagnostic methods are being integrated into lab-on-a-chip platforms that utilize microfluidic and biosensor technologies and are emerging as ideal platforms for POC diagnostics. Separate nucleic acid amplification steps increase the assay time and complexity and introduce a cross-contamination risk during sample transfer to the CRISPR reaction. Therefore, integrating CRISPR assay procedures into user-friendly devices, such as microfluidic chips or lateral flow assays, allows for the provision of qualitative and quantitative readouts [30].
In this study, cDNA was synthesized using primers designed for BVDV-1b obtained from the Korea Veterinary Culture Collection (KVCC). The crRNAs were designed to have high specificity for the target pathogen. The Cas13 enzyme was expressed and purified. For detection, the BVDV-1b target cDNA, crRNA, Cas13 enzyme, and RNA reporter were subjected to collateral cleavage reaction at 37°C. Lateral flow cytometry analysis showed that crRNAs 1, 2, 3, and 4 were positive for BVDV-1b. If CRISPR-Cas13-based detection is conducted, BVDV-1b can be rapidly and accurately diagnosed, even in samples collected under harsh conditions with a high risk of contamination. We propose that our results could serve as a new POC test at the DNA level for detecting nucleic acid of the BVDV-1b, which frequently occurs in the Republic of Korea. In the next study, we will conduct experiments using clinical samples.