













INTRODUCTION
Livestock plays an integral part in human civilization in terms of the early drafting performance and long-term supply of protein including meat, egg and dairy. High harvest is one principal objective for livestock production, leading to persistently intense artificial selection for economic traits, namely selective breeding. Only a few excellent individuals inevitably have an opportunity to generate offspring, especially the sire considering the maintaining cost and popularization of artificial insemination, which contributes to the decline of genetic diversity at the population and individual levels. Consequently, the phenomenon inbreeding, leading to increased homozygosity of progeny, always occurs in livestock breed, coupled with the reduction in evolutionary fitness of the next generation referred to as inbreeding depression [1]. As a result of inbreeding depression, it is hard to maintain superior performance, even normal fitness for individuals. More attention was poured to the selection on growth performance and reproductive traits in livestock production, and it is worth noting the potentially negative effects of inbreeding depression on these traits. Therefore, the pros and cons of selective inbreeding are apparent, where one is the improvement of productive performance, and another is the risk of inbreeding depression. It has been reported that inbreeding depression occurs in numerous animals, including pigs [2], cattle [3], horses [4], sheep [5], goats [6], chickens [7] and dogs [8]. Hence, crucial to the effective breeding management is the regularly genetic monitoring and assessment of inbreeding.
The classic method to evaluate inbreeding depression is based on fitness-related traits from individuals with grouped inbreeding, which is measured by the probability of inheriting two alleles identical-by-descent at an autosomal locus in the presence of consanguinity, with so-called inbreeding coefficient [1,9]. Traditionally, inbreeding coefficient was calculated based upon pedigrees involved in thousands of individuals; however, recent estimates of inbreeding coefficient relying on genomic data were recommended for the sake of accuracy and convenience without the relatives of subjects [10].
Apart from artificial selection, natural selection also acts in farm animals to adapt various environments via three ways (positive selection, background selection and balance selection) [11]. Both selections can shape individual genome, resulting in genomic footprints of selection called signatures of selection, of which the whole-genome scan can return genetic information to disclose possibly genetic makeup of adaptive evolution, to identify promising genes responsible for importantly agronomic traits, to estimate the development of herds, and to provide some management strategies [12]. The emergence of a plethora of algorithms and software for detecting selective sweeps provides an opportunity to decipher the genetic architecture of given phenotypes.
Intense artificial selection has been imposed to Luzhong mutton sheep population in the past years. Improvements on growth and reproductive performance are two breeding goals in the present herd. Although some progresses were phenotypically observed compared to the unselected population (F2), such as birth weight (4.04 ± 0.06 vs 3.36 ± 0.03 kg, p < 0.05, t-test), and average litter size (1.91 ± 0.07 vs 1.37 ± 0.03, p < 0.05, Mann-Whitney U-test), the genomic evaluation was poorly understood. Therefore, the objectives of this study were to characterize the pattern of runs of homozygosity (ROH), estimate the effects of inbreeding on early growth and productive performance, and detect signatures of positive selection based upon haplotypes in Luzhong mutton sheep.
MATERIALS AND METHODS
In total, the blood samples of 345 Luzhong mutton sheep under intensive feeding were collected from Ji’nan Laiwu Yingtai Agriculture and Animal Husbandry Technology (N36°17′33.25″ E117°35′14.98″, Ji’ nan, Shandong, China) for extraction of genomic DNA using a QIAamp DNA Blood Midi Kit (Qiagen, Germany), of which 277 animals harbored records of birth weight and body conformation traits at birth including body length, chest girth and withers height, and 70 individuals held reproductive performance (specifically, litter size) of at least two parities. Each 15 individuals were kept in one pen with 3 m2/animal, and all ewes were ~3.5 kg fed silage maize, ~1 kg concentrate feed mixture, and ad libitum water. All animals were genotyped by a high-density SNP (single nucleotide polymorphism) chip featured by 633,619 markers, followed by quality control using PLINK v1.90 [13] with criteria: (1) call rate ≥ 90%; (2) minor allele frequency ≥ 1%; (3) p-value of Hard–Weinberg disequilibrium test ≥ 10−6; (4) sample call rate ≥ 95%; and (5) markers on autosomes. The data set used in this study were deposited in Figshare (https://doi.org/10.6084/m9.figshare.9987965, and https://doi.org/10.6084/m9.figshare.12052824), partly accessible in previous work [14].
ROH calling was performed by PLINK v1.90 [13] with following parameters: (1) length of ROH ≥ 500 kb; (2) number of SNPs within ROH ≥ 100; (3) density of marker ≥ one SNP per 50 kb; and (4) the gap between proximal two SNPs ≤ 1,000 kb. One heterozygote and five missing calls were allowed within the sliding window of 5,000 kb. Moreover, “--homozyg-group” command in PLINK v1.90 [13] was executed to identify homozygous identical segments among individuals referenced as to ROH hotspots, which was defined as the segments with length over 1 kb and shared by at least 173 animals [(345 + 1) / 2 = 173]. Genomic inbreeding coefficients based on ROH (FROH) were estimated by the proportion of the total length of ROH on all autosomes (2,655.71 Mb used in this study).
All animals were divided into three categories based on FROH, including FROH < 0.01, 0.01 ≤ FROH ≤ 0.1 and FROH ≥ 0.1. For early growth performance traits, we investigated the effects of genomic inbreeding coefficient categories, taking sex as a fixed factor in the model. Tukey method was used to paired comparison, declaring significance at p level of 0.05. For litter size of the first and second parity, average litter size of the first two parities, and average litter size, one-way ANOVA was employed to explore the effect of inbreeding coefficients, accepting significance of 0.05. Moreover, we regressed both early growth and reproduction performance for each sheep against FROH to assess inbreeding depression.
To investigate the genomic footprints left by selection in the present population, the statistic number of segregating sites by length (nSL) [15], a log-ratio of the SL statistic calculated for the ancestral and derived haplotype pools, was employed to detect selective sweeps considering soft sweeps. Haplotypes were phased severally for autosomes using shapeit v2.12, followed by nSL calculation by selscan v1.2.0, and normalization with norm [16,17]. Top 5% loci were considered as candidates and annotation to sheep reference genome Oar_v4.0 was performed by Variant Annotation Integrator online. DAVID (accessed April 19, 2020), STRING (accessed April 22, 2020) and Animal QTLdb (accessed April 22, 2020) database were used for functional annotation and interactions of candidate genes.
RESULTS
Overall, sample genotyping rate was 98.34%. As a consequence of quality control, 522,102 SNPs and 345 samples passed filters, during which 30,061 variants were removed due to missing genotype data, 9,648 Hardy-Weinberg exact test, and 71,808 minor allele frequency.
A total of 22,821 ROHs were found in the present population. At the individual level, the number of ROH ranged from 1 to 170, the total length 532 kb to 745,382 kb, and the average length 532 kb to 8,191 kb (Fig. 1A). According to the length, all ROHs were grouped into three classes, showing a large proportion were short (< 2 Mb) (Fig. 1B).

In total, ten ROH hotspots were identified, including three on chromosome 1 and seven on chromosome 10 (Table 1). None gene was found on seven ROH hotspots on chromosome 10, whereas two genes were discovered on three ROH hotspots on chromosome 1, containing STAG1 (stromal antigen 1) involved in cell cycle and PCCB (propionyl-CoA carboxylase subunit beta) participating in metabolism.
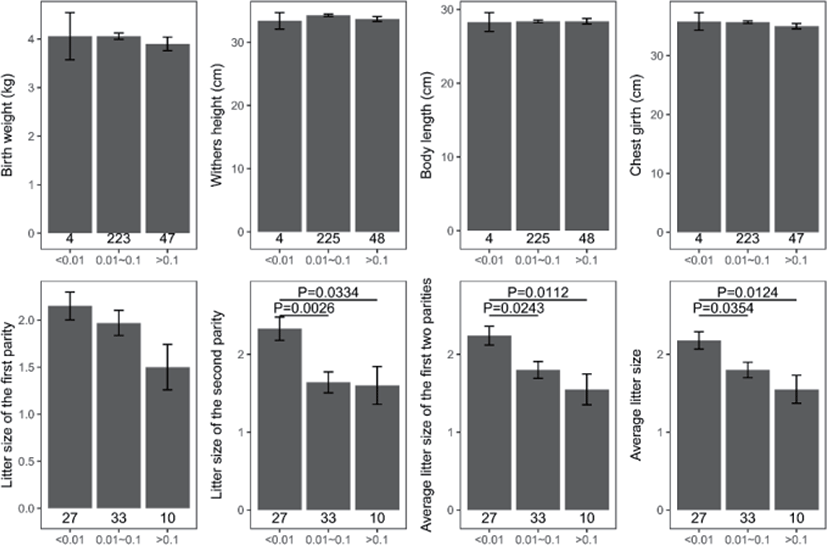
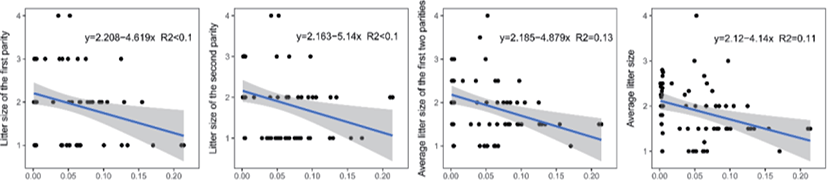
Genomic inbreeding coefficients FROH ranged from 0.0002 to 0.2807, with an average of 0.0649, indicating a low level of inbreeding in the present population. There were no significant differences between the category of genomic inbreeding coefficients (FROH < 0.01, 0.01 ≤ FROH ≤ 0.1 and FROH > 0.1) in terms of early growth performance traits, harboring birth weight and body conformation at birth (withers height, body length and chest girth) (Fig. 2). However, compared to ewes with low inbreeding coefficients (FROH < 0.01), remarkable diminishes of litter size were observed for these with high inbreeding except the first parity (p < 0.05), implying the presence of inbreeding depression on reproductive performance in this population (Fig. 2). Moreover, linear regression analysis of birth weight, body conformation at birth and litter size was performed to explore the effects of inbreeding on early growth performance and reproductive capacity. As a consequence, albeit the absence of valid regression for early growth performance and the presence of poor fitting degree for litter size, a trend of decline for litter size was observed based on negative regression coefficients, suggesting potential inbreeding depression on both reproductive traits in Luzhong mutton sheep (Fig. 3).
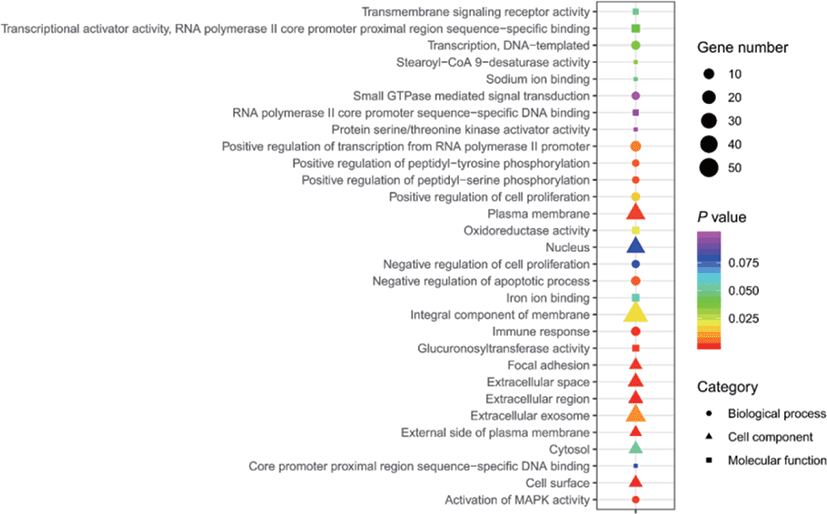
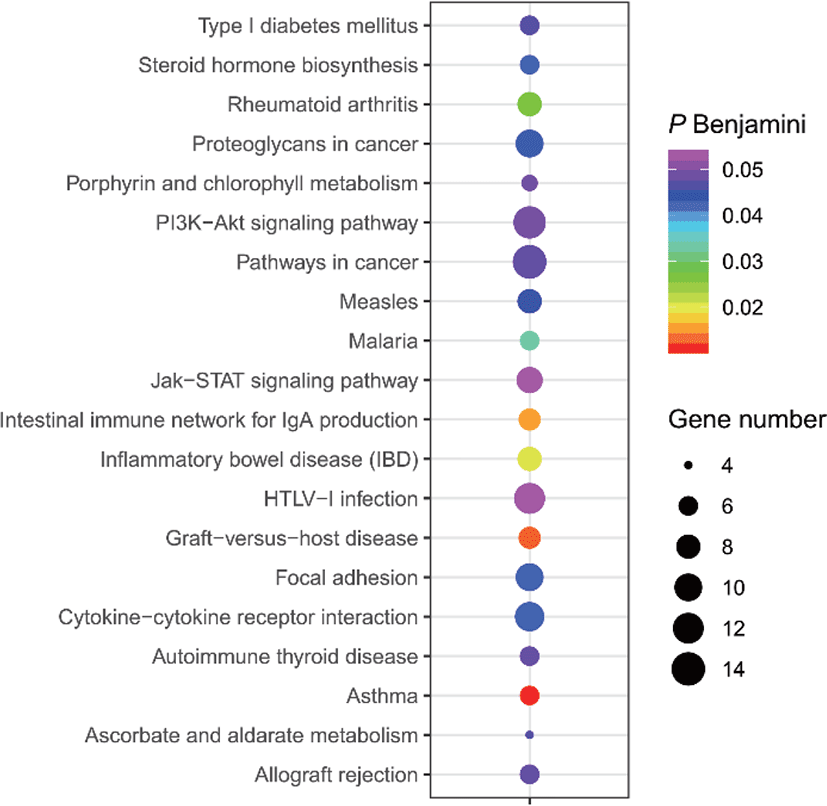
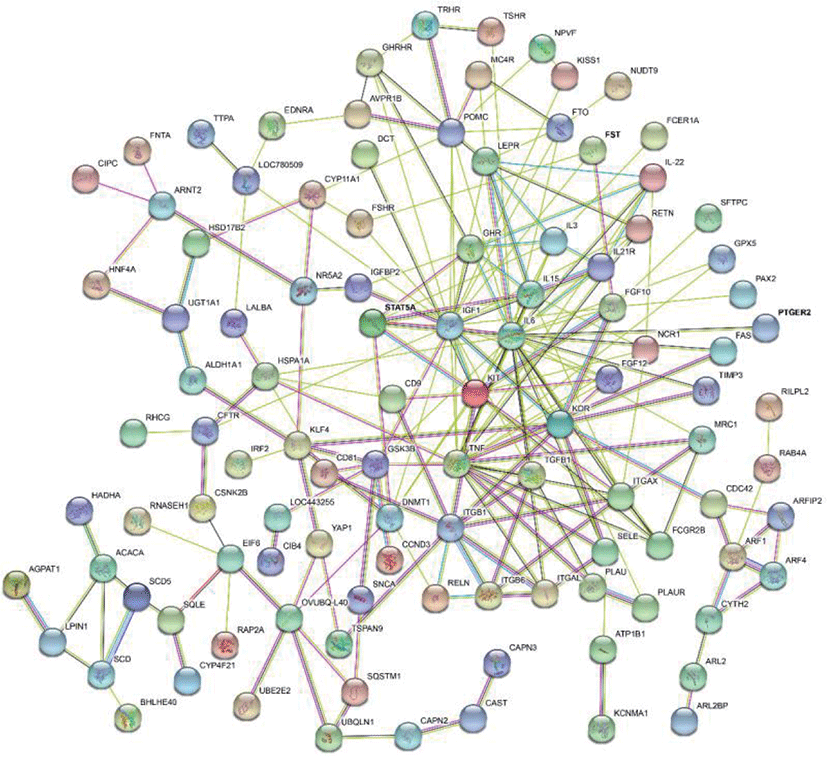
The results of selective sweeps were shown in Fig. 4. A total of 160 candidate genes were identified for the top 5% loci, which were enriched to 87 gene ontology (GO) terms including 59 biological process terms, 18 cell component terms and ten molecular function terms, and 52 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, respectively. Of these genes, signal transducer and activator of transcription 5A (STAT5A) with extensive functions was found with highest |nSL|, followed by two hormone-related genes follistatin (FST) on chromosome 16 and PTGER2 (prostaglandin E receptor 2) on chromosome 7. Extensive and fundamental roles in cell component and molecular function were observed for most candidate genes under selection (Fig. 5). These genes were involved in some important biological processes such as regulation of cell proliferation (GO:0043066, GO:0008284 and GO:0008285), immune response (GO:0006955), regulation of protein function (GO:0050731 and GO:0033138), signal transduction (GO:0000187 and GO:0007264), and biochemical function (GO:0045944 and GO:0006351) (Fig. 5). Significant KEGG pathways include numerous diseases, immunoregulation, metabolism, signaling pathways, and steroid hormone biosynthesis (Fig. 6). To explore the relationship between candidate genes, the primary interactions network was represented in Fig. 7. In total, 19 genes were found in Animal QTLdb for several traits, including growth and development, reproduction, carcass and meat quality, milk composition, fiber quality, and parasite/diseases resistance (Table 2).
1) This table is based on the information searched in Animal QTLdb (accessed April 22, 2020) database.
GHR, growth hormone receptor; MB, myoglobin; LEP, leptin receptor; GHRHR, growth hormone releasing hormone receptor; CAST, calpastatin; FAS, Fas cell surface death receptor; LALBA, lactalbumin alpha; FSHR, follicle stimulating hormone receptor; TGFB1, transforming growth factor beta 1; ACACA, acetyl-CoA carboxylase alpha; SCD5, stearoyl-CoA desaturase 5; ITGB1, integrin subunit beta 1; IGF1, insulin like growth factor 1; STAT5A, signal transducer and activator of transcription 5A; FST, follistatin; KCNMA1, potassium calcium-activated channel subfamily M alpha 1; IL6, interleukin 6.
DISCUSSION
High harvest is one principal goal of selective breeding in livestock, during which phenotypical and genetic evaluation are always needed to monitor the achievement of breeding. Here, we evaluated the genomic inbreeding grounded on homozygosity and its effects on early growth performance and litter size, detected positive selection signatures based upon haplotype using a high-density SNP array, which may provide some useful information for breeding progress on Luzhong mutton sheep.
There was a long time when genetic evaluation for farm animals depended on pedigree data involved in thousands of individuals from several decades, which frequently contained parentage errors. Fortunately, genomic data provide an opportunity to get accurate and convenient assessments for animals in a lack of pedigrees. Inbreeding is a good example in this point in that some genetic inbreeding coefficients have been developed, of which FROH, the proportion of total ROH length on genomic autosomes, has been fully recognized. Inbreeding, mating between related animals, increases the genomic homozygosity of offspring. Longer ROH are expected in individuals with higher inbreeding degrees. We found a large proportion of short ROH (< 2 Mb), implying a low inbreeding degreed in our population. It also was demonstrated by a low estimator of inbreeding coefficients (on average 0.0649). Indeed, this population is a new line with a relatively short breeding history. Hence, it was unsurprising that inbreeding depressions were not observed on early growth performance. Albeit low inbreeding and small sample size, inbreeding depressions on litter size were detected in this study, probably due to the accurate estimator of inbreeding coefficients. Some hypotheses have been developed for inbreeding depression that it is caused by homozygosity of rare lethal, homozygosity of variants maintained by overdominance, nearly recessive variants and partially recessive, detrimental variants [9,18,19]. One possible explanation to the inbreeding depressions on litter size in low inbreeding population is that some major detrimental loci have been recessive homozygous, implying the importance and urgency of identifying these loci. Avoiding the occurrence of inbreeding depression is one approach to maintain the performance of herds. To reduce high inbreeding depression, minimizing inbreeding or mean kinship by reducing the weight given to family information at the selection step or optimal contribution selection, purging induced by deliberate inbreeding and genetic rescue (called hybridization in farm animals) can be implemented to manage a population [9]. Considering the low and slow inbreeding level in the current population, purging is effective to reduce or eliminate detrimental variants. To do this, following with subdividing the population into isolated lines, between-lines selection can increase purging [20], where the caveat is that elevated genetic drift will reduce overall genetic diversity. Alternatively, another strategy is to induce inbred mating, not increasing drift [21].
Improvement on growth performance is one of the breeding objectives of Luzhong mutton sheep population. Expectedly, several genes linked to body weight were identified by selective sweep analysis, such as growth hormone receptor (GHR), insulin-like growth factor 1 (IGF1), growth hormone releasing hormone receptor (GHRHR) and leptin receptor (LEPR). The growth hormone (GH) was known to modulate growth via the GH-GHR-IGF1 axis [22]. GH promotes body growth by binding dimerized GHRs to form a trimolecular complex, inducing downstream signaling pathways such as Janus kinase 2 (JAK2) activation, STATs, SRC family kinase (SFK), and JAK–STAT signaling [23]. Interestingly, the strongest selection signature was identified on STAT5A, which plays a vital role in GH-GHR-IGF1 axis [23,24]. Skeletal growth and mineral acquisition were regulated by GHR by the GH/IGF1 axis in mice [25]. Polymorphisms on GHR, GHRHR, and IGF1 were found to be associated with milk yield and quality traits in Sarda sheep [26], indicating the roles of these genes in controlling offspring’s early growth through nutritional regulation. LEPR is the receptor of leptin which is a hormone synthesized by adipocytes and influences development, growth, metabolism and reproduction [27]. It is important to note that leptin binds to LEPR, followed by downstream JAK2 activation and STAT3 signaling [28,29], suggesting the potential interaction between LEPR and GH-GHR-IGF1 axis.
Another breeding objective of the present population is to increase litter size by marker-assisted selection of FecB BB genotype on BMPR1B (bone morphogenetic protein receptor type 1B). Although known BMPR1B did not emerge from selective sweep, several related genes including follicle-stimulating hormon receptor (FSHR), GHR and FST were found to be under selection in this study. Polymorphisms of FSHR were reported to be associated with litter size of some prolific breeds including Small Tail Han, Hu, Hetian and Bashbai sheep [30–33]. It is also important to note that GHR has been identified as a candidate gene for litter size of Texel sheep in a genome-wide associated study [34]. FSHR and GHR, the receptor of follicle-stimulating hormone and growth hormone, interact with BMPR1B to act as important regulators in the development of follicle. Interestingly, the patterns of DNA methylation on BMPR1B, FST and FSHR may participate in the regulation of ovine fecundity [35]. Folliculogenesis was modulated by pituitary gonadotrophins and intraovarian growth factors including epidermal growth factor (EGF), fibroblast growth factor (FGF), transforming growth factor-α (TGF-α), TGF-β, IGFs, IGF-binding proteins (IGFBPs), growth differentiation factor 9 (GDF-9), BMPs, inhibin, activin and FST [36], of which some genes were under selection in our study. It is interesting to note that FST, coding FST, was identified with top 2 |nSL| value. The expression location of FST mRNA and protein within ovary is granulosa cell in antral follicles and luteinized granulosa cell [37], suggesting the role of FST in regulating follicular development. FST is a binding protein belonging to the TGF-β superfamily, acting as an inhibitory by binding to other members of the TGF-β superfamily including activin, BMP-4, BMP-7, BMP-15, and GDF-8, which play curial roles in ovarian function [36]. The increased FST in ovary decreased the number of FSH and LH receptors in granulosa cells, progesterone level in undifferentiated granulosa cells and estrogen level, and increased progesterone level in differentiated granulosa cells and androgen, leading to the suppression of oocytes in terms of meiotic and cytoplasmic maturation, and developmental competence to form blastocysts [36]. Together, genes associated with litter size have been under selection, suggesting the effectiveness of selective breeding on litter size in Luzhong mutton sheep. However, more work is needed to disclose potential causative variations for litter size in the current population.
To sum up, we estimated genomic inbreeding and its effects on early growth and reproductive performance based on whole-genome homozygosity, and detected selection signatures based upon haplotypes to evaluate the breeding progresses on the recent herd. Accordingly, purging selection was proposed to alleviate the inbreeding depression on litter size. Selective sweeps on genes associated with growth and litter size highlighted the effectiveness of the persistent selective breeding in Luzhong mutton sheep population. These results provide novel insights to evaluate breeding progresses on livestock, and are in favor in genetic improvement of the present flock.