













INTRODUCTION
Globally, per capita meat consumption and total meat consumption are increasing as economies grow and populations increase [1]. The current world population is approximately 7.8 billion, and it is projected to increase to 9.6 billion by 2050 [2]. As a result, it has reached a situation where food and meat production must be further increased. However, in recent years, there has been a growing notion that conventional meat production via livestock has high greenhouse gas emissions, energy, and land use [3,4]. Thus, alternative ways of meat production are needed. As an alternative, cultured meat is being studied with the aim of solving problems related to animal welfare and sustainability issues [5]. Important elements of cultured meat production technology include cells, animal-derived and synthetic materials, culture media, scaffolds, which mimic the in vivo muscle-generating environment (humidity, temperature, air composition, dissolved oxygen tension, etc.) [6,7]. The cultivation temperature and oxygen concentration have been reported to have a significant impact on the physiological activity of cells [8–11]. Heat stress is a state that happen when a live-stock is unable to expel heat produced or absorbed by the body to maintain internal heat balance [12]. In living cells, heat stress is one of the environmental stresses that can negatively affect and change cell metabolism. It can inhibit protein synthesis, change protein folding and function, and change metabolism and membrane fluidity [13–15]. However, depending on the intensity and duration of applied heat, its effects on muscle cell activity are different [16]. Mild heat stress can increase mitochondrial biogenesis in cells with a positive effect on mitochondrial oxidative phosphorylation in skeletal muscle [17,18]. Increased mitochondrial biogenesis ultimately produces adenosine triphosphate (ATP) by enhancing mitochondrial oxidative phosphorylation [19]. In addition, mild heat stress promotes cell proliferation and differentiation [2,20]. Oxygen and glucose are important for cellular respiration because aerobic respiration during glycolysis provides more ATP per glucose molecule than anaerobic respiration [21]. In addition, cells not only can utilize oxygen for ATP resynthesis, but also can detect oxygen levels and respond to low oxygen concentration with adaptations regulated by signaling proteins such as hypoxia-inducible factors [22]. The average concentration of physiological oxygen in skeletal muscle is about 2.3% to 6.0% [23]. Previous studies have reported that culturing muscle cells in a hypoxic environment similar to physiological oxygen concentrations enhances proliferation [2,24–28]. However, the effect of interactively applying heat stress and hypoxia on production capacity of cells for cell culture meat production has been hardly studied. Therefore, this study aimed to analyze the impact of heat stress and hypoxic conditions on the proliferation and differentiation of muscle cells.
MATERIALS AND METHODS
C2C12 cells used in the experiment were purchased from ATCC® (CRL-1772TM 79). All experiments used Dulbecco’s modified eagle’s medium (DMEM; Gibco, Waltham, MA, USA) as the culture medium for cell growth, to which 10% of fetal bovine serum (FBS; Corning, Corning, NY, USA) and 1% of Penicillin-Streptomycin-Amphotericin B Mixture (PSA; Lonza, Basel, Swiss) were added. For the differentiation medium, DMEM supplemented with 1% PSA and 2% FBS was used. In the case of proliferation during cell experiments, 1,800 cells per cm2 T25-flask were seeded. Cells were cultured in incubators under the following conditions for 5 days: CON (37°C, 20% O2), T1 (37°C, 2% O2), T2 (39°C, 20% O2), and T3 (39°C, 2% O2). CO2 concentration was the same at 5%. Oxygen concentration was adjusted using N2 gas. In the case of differentiation, 1,800 cells per cm2 T25-flask were seeded and cultured in the same conditions as proliferation conditions. When the cells in the flask became confluent, the growth medium was aspirated by suction and washed with phosphate buffered saline solution (PBS; SH30256.01, Cytiva, Marlborough, MA, USA). Then proceed with differentiation by replacing with differentiation medium.
The flask used for all culture experiments was used after coating to facilitate cell attachment and growth. Collagen coating solution was manufactured by diluting 1M acetic acid (A0052, Samchun chemicals, Seoul, Korea) in distilled water to a concentration of 2% of the solution and then adding a 5 mg/mL collagen solution (A1064401, Gibco) to a concentration of 0.5% to solubilize the collagen. After the collagen solution was dispensed into the flask. Before using in the experiment, the coating solution was removed by suction and the flask was washed twice with PBS. Matrigel coating solution was manufactured by diluting Matrigel solution in cold PBS to a concentration of 0.05% of the solution. Before using it in the experiment, the coating solution was removed by suction and the flask was washed with PBS.
Cell count assay and MTS assay were performed when cells became confluent (5 days). Attached cells were added with 0.05% trypsin-EDTA (25300-062, Gibco) for 3 minutes. Using a pipette, content of each flask was moved to a 50 ml tube and centrifuged at 352×g for 5 minutes using a centrifuge. Suctioned out the supernatant, added media to the pelleted cells, and stained the cells with Trypan Blue (T8154, Sigma-Aldrich, St. Louis, MO, USA) The number of cells was counted using a cell counter (AMQAX2000, Invitrogen, Carlsbad, CA, USA). MTS assay was performed using C2C12 cells cultured in a 96-well plate. After removing culture medium, MTS reagent (G3582, Promega, Madison, WI, USA) was then dispensed into each well and absorbance was measured at 490 nm.
Proliferation proceeded on the 5th day and differentiation proceeded on the 3rd day. After aspirating the medium, 2% paraformaldehyde (SM-P01-050, GeneAll Biotechnology, Seoul, Korea) was added and incubated at 37°C for 45 minutes. Washed twice with PBS. And it was incubated at room temperature for 20 minutes using 0.1% Triton X (T8787, Sigma-Aldrich). They were then incubated for 30 minutes in 2% bovine serum albumin (BSA;10738328103, Roche, Basel, Swiss) at room temperature. Then, the flask was washed with PBS. For proliferation, cells were treated with anti-Pax7. For differentiation, cells were treated with anti-MyoD, anti-Myosin and Anti-Desmin followed by incubation at 4°C overnight. Washed twice with 0.05% Tween 20 and incubated with secondary antibody Alexa Fluor 488 (A21121, Invitrogen) and finally Hoechst 33342 reagent (H3570, Invitrogen). Thereafter, after washing twice with 0.05% Tween 20 (1706531, Bio-rad, Hercules, CA, USA), immunofluorescence-stained C2C12 myoblast cells were observed using an microscope (EVOS-5000, Invitrogen). For image data, the total number of nuclei, the number of cell nuclei in myotube and the fusion index were calculated using Image J program (NIH) by a skilled expert.
To cool the T25 flask, the medium was aspirated on ice and washed once with PBS. After adding radioimmunoprecipitation assay (RIPA) lysis buffer (MB-030-0050, Rockland, Pottstown, PA, USA) to the flask, the cells were separated with a cell scraper. Extracted proteins were quantified by Bradford assay. Extracted proteins were separated by electrophoresis on a gel (4561024, Bio-rad). Proteins in the gel were transferred to membranes (1620175, Bio-rad). Membranes were blocked by incubation in blocking buffer (EveryBlot, Bio-rad) at room temperature for 1h. Incubate overnight at 4°C with primary antibodies to measure Myh, Myod1, Myog, Mb and Actb. Membranes were then washed with Tris-Buffered Saline (1706435, Bio-rad) with 0.1% Tween 20 (1706531, Bio-rad) (TBST) 4 times (10 minutes each time) and incubated with Affinity Purified Goat Anti-Mouse IgG (H+L) Horseradish peroxidase (HRP)-conjugated or Affinity Purified Goat HRP-conjugated Anti-Rabbit IgG (H+L) for 1h at room temperature. After washing again with TBST 4 times (10 min each time), Clarity western ECL substrate (1705061, Bio-rad) was dispensed to detect proteins. After detection, Western Blot Stripping Buffer (21059, Thermo Scientific, Waltham, MA, USA) was dispensed to treat membrane for 1 hour. Membranes were then stored, or proteins were detected by performing the same process again using blocking buffer.
mRNA was conducted on cells from the 3 day of differentiation, extracted using Total RNA Extraction Kit (iNtRON Biotechnology, Seongnam, Korea). mRNA was converted to cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA). mRNA was converted to cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) as followed: 25°C for 10 min, at 37°C for 2 h, and at 85°C for 5 min. Gene amplification was performed using Fast qPCR 2×SYBR Green Master Mix (Applied Biosystems). Gene amplification was performed for 40 cycles as followed cycle: 50°C for 2 min and 95°C for 10 min; 15 secs at 95°C; 1 min at 53°C; 15 secs at 95°C; 1 min at 53°C. The target gene were Myod1, Mb, Myog,Myh2 and Gapdh. Relative gene expression was calculated using the 2−ΔΔCt method [28]. Primers used for analysis were Myod1 (forward [F]) 5’-AGCTTCTGTCTCCCTTCATC-3’ and (reverse [R]) 5’-CACAAC CAAGGTCCTATTCA-3’; Mb (F) 5’-GGAAGTCCTCATCGGTCTGT-3’ and (R) 5’-GCCCTTCATATCTTCCTCTGA-3’; Myh2 (F) 5’-GAAAAAGAAGGAGG AGGTGA-3’ and (R) 5’-CCAAAGTGAATCCTGATGAA-3’; Myog (F) 5’-GCCCAG TGAATGCAACTCCCACA-3’ and (R) 5’-CAGCCGCGAGCAAATGATCTCCT-3’; Gapdh (F) 5’-GTGGCAAAGTGGAGATTGTTGCC-3’ and (R) 5’-GATGATGAC CCGTTTGGCTCC-3’.
All data were tested by two-way Analysis of variance (ANOVA). All data were statistically verified for statistical significance using the statistical processing program SAS (SAS Institute, Cary, NC, USA). Duncan’s multiple range test was used to compare significant differences in measured values and a p-value of less than 0.05 was considered statistically significant.
RESULTS
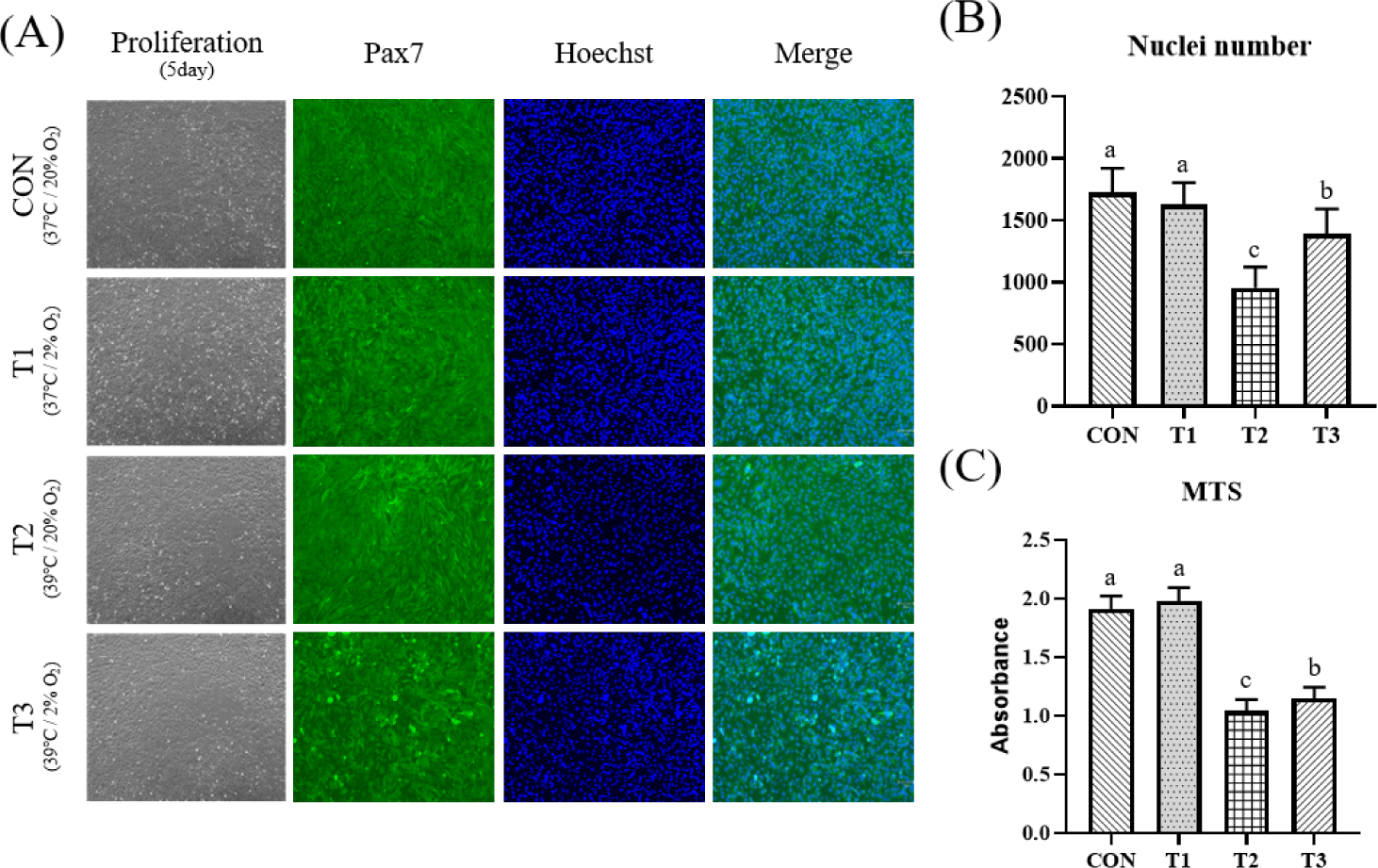
To investigate effects of temperature and oxygen concentration conditions on muscle cell proliferation, C2C12 cells were grown under different temperatures (37°C, 39°C) and oxygen concentrations (20%, 2%) for 5 days. Table 1 shows cell count results of C2C12 according to temperature and oxygen concentration. Total cell number and live cell number were significantly higher in CON and T1 under a temperature condition of 37°C than those in T2 and T3 under a temperature condition of 39°C (p < 0.05). As shown in Fig. 1, MTS assay and immunofluorescence staining were performed to confirm proliferative capacity. Absorbance value of the MTS assay indicates mitochondrial activity of cells, which was significantly higher in CON and T1 (p < 0.05). The number of nuclei obtained by immunofluorescence staining was also significantly higher in CON and T1 (p < 0.05). Table 2 shows influence of each condition and interaction through two way-ANOVA statistics for all experimental results. In the case of temperature, it affected total cell number (p < 0.05), live cell number (p < 0.05), MTS absorbance (p < 0.0001), and nuclei number (p < 0.0001), whereas oxygen concentration condition had little effect.
Next, effects of temperature and oxygen concentration on differentiation were investigated. After C2C12 cells became confluent under the same temperature and oxygen conditions, they were differentiated for 3 days at different temperature (37°C, 39°C) and oxygen concentration (20%, 2%) conditions. Resulting myotubes were fixed, immunofluorescence stained for Myosin and Desmin, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Plates were imaged and myogenesis was quantified by measured the number of nuclei in myotube and the fusion index. In addition, relative mRNA expression levels of Myog, Mb, Myh2, and Myod1 as important genes for muscles were measured using RT-qPCR. Protein expression levels of Myh, Myod1, Myog and Mb were measured by Western blotting. Fig. 2 shows results of immunofluorescence staining, number of myotube nuclei, and fusion index after differentiation of C2C12 cells. Based on the photograph of Fig. 2A, numbers of myotube nuclei and fusion index were measured. Myotube nuclei showed no significant difference (p > 0.05). The fusion index showed a significantly higher value at T3 than at T1 (p < 0.05). In Table 2, myotube area had a significant effect at the temperature and oxygen conditions (p < 0.05). The fusion index tended to be affected by temperature conditions and interaction conditions (Tem × Oxy) (p < 0.1) (Table 2).
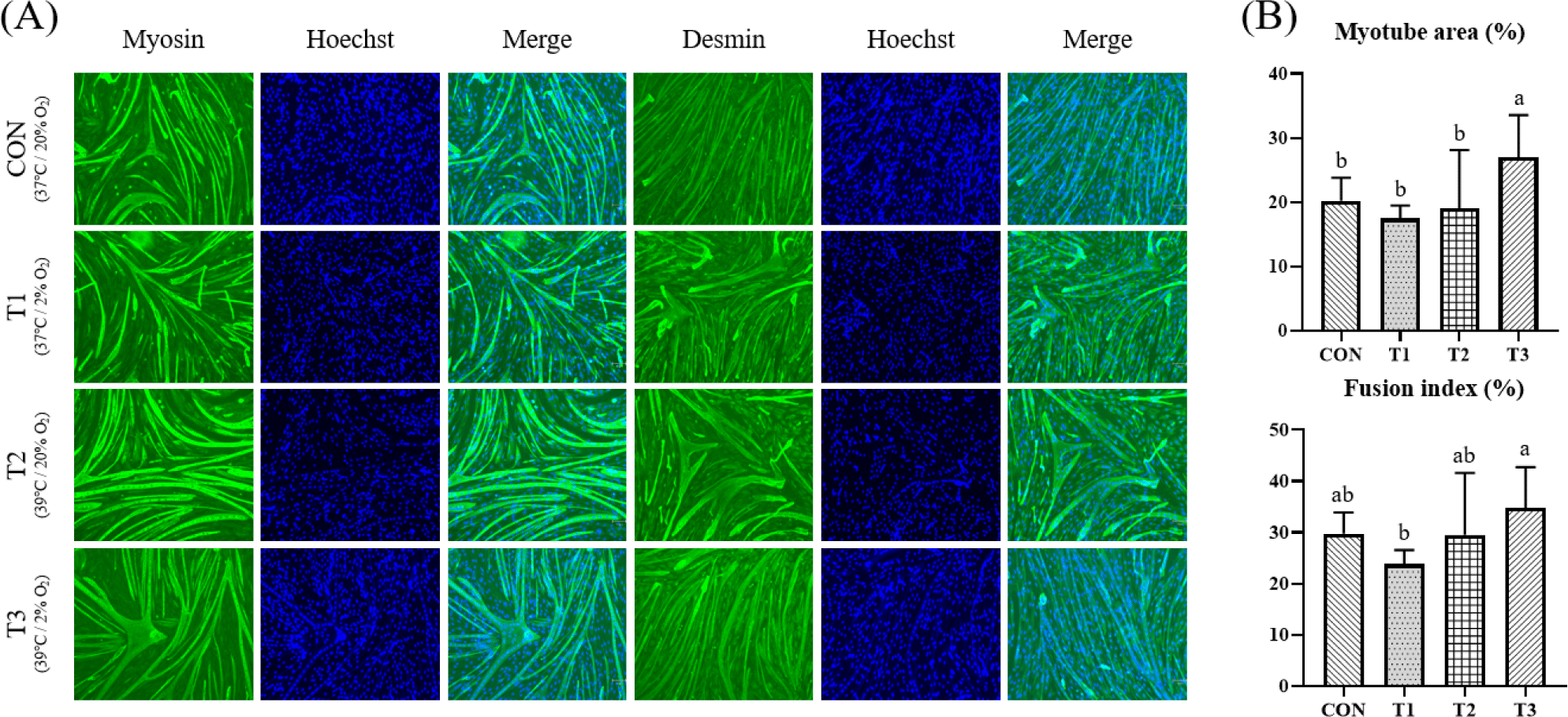
Fig. 3 shows results of relative mRNA expression levels of Myog, Mb, Myh2, and Myod1 using RT-qPCR and Western blotting pictures of myotube formation on day 3. In the case of Myog, it showed the highest value in T3. Its expression level in T3 was significantly higher than that in T1 or T3 with 2% oxygen concentration or that in CON or T2 with 20% oxygen concentration (p < 0.05). Mb and Myh2 levels were the highest in T3. Their expression levels were significantly higher when the temperature was 39°C than those when the temperature was 37°C (p < 0.05). In the case of Myod1, it had a significantly higher value in T3 than in CON (p < 0.05). Temperature conditions significantly affected expression levels of Myog (p < 0.05), Mb (p < 0.0001), and Myh2 (p < 0.0001). Oxygen concentration significantly affected Myog (p < 0.001), and Myod1 (p < 0.05) (Table 2).
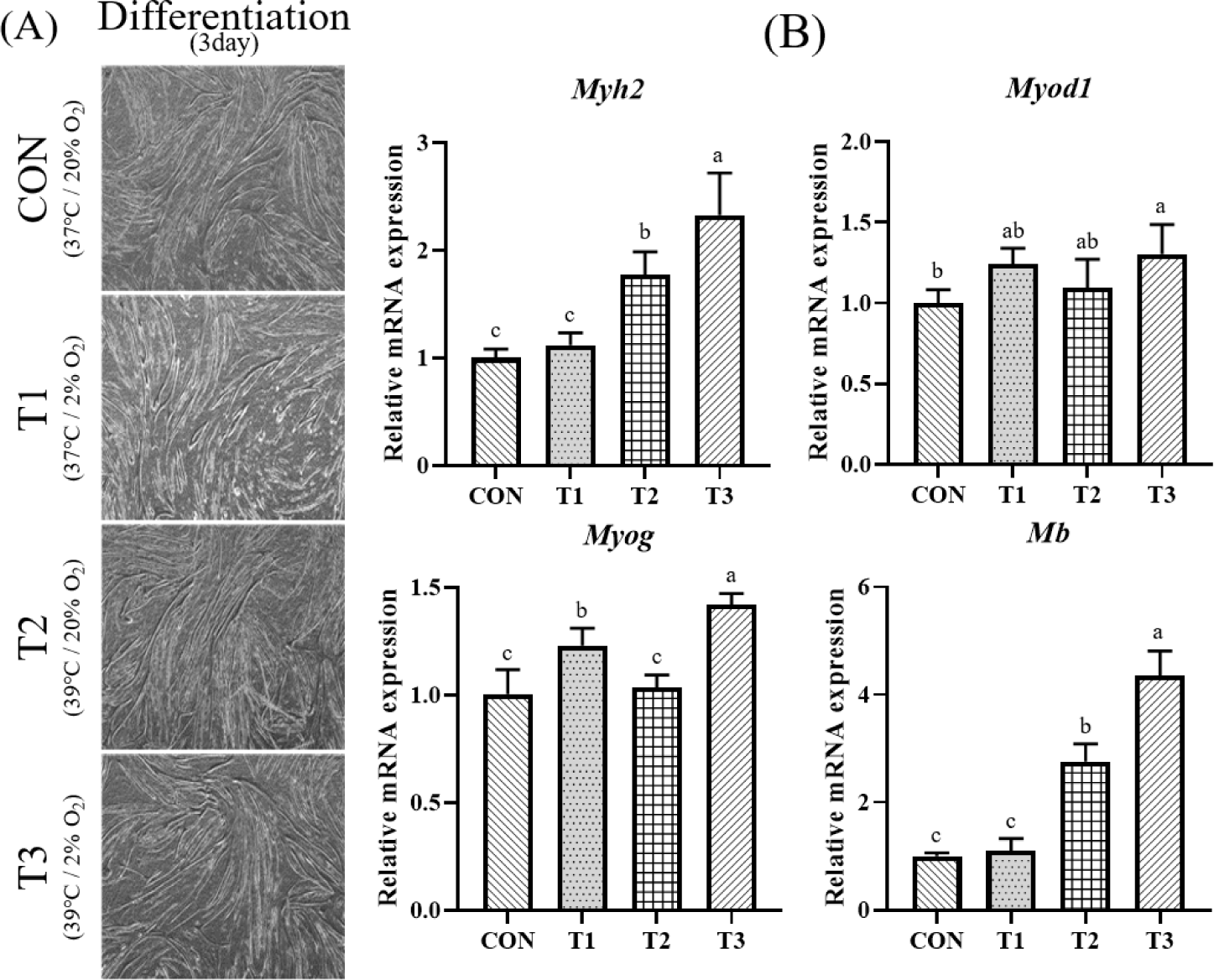
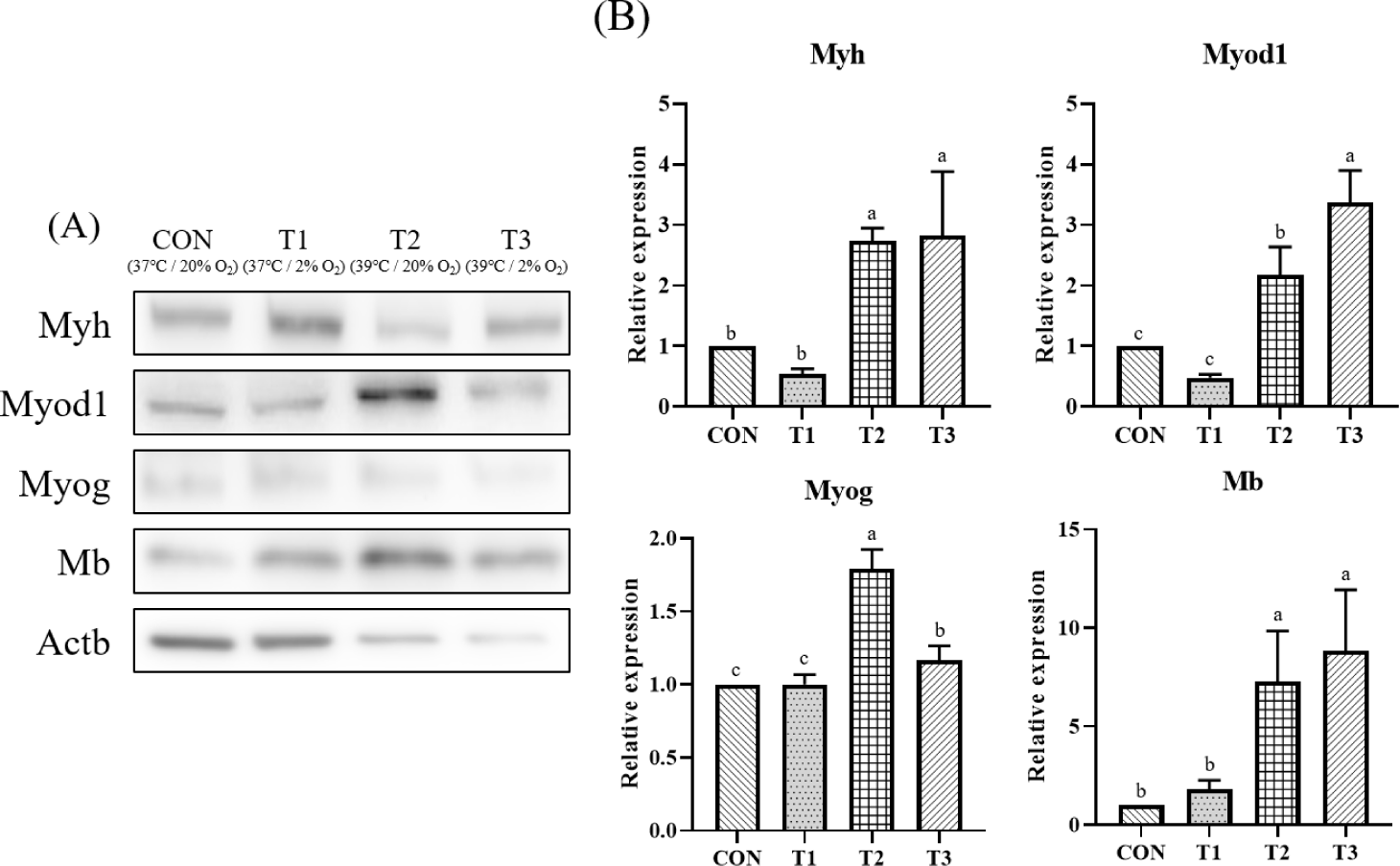
Fig. 4A is a photograph of fluorescence intensity measured by Western blot for Myh, Myod1, Myog, Mb and Actb. Fig. 4B shows results of fold change for CON by normalizing fluorescence intensity with beta-actin, a housekeeping gene. Expression of Myh was significantly higher in T3 than in CON (p < 0.05). Its expression was higher at 39°C than that at 37°C. Expression of Myod1 was significantly higher in T2 and T3 when the culture temperature was 39°C. But there was no significant difference between treatment groups at the same culture temperature and different oxygen concentrations (p < 0.05). Expression of Myog was significantly higher in T2 and T3 at 39°C than in CON and T1 at 37°C (p < 0.05). When the culture temperature was the same with different oxygen concentration, significantly lower values were found at 2% in T1 and T3 than at 20% in CON and T2. In the case of Mb, there was no significant difference according to oxygen concentration. However, T2 and T3 treatment groups with a culture temperature of 39°C showed significantly higher expression levels than CON and T1 with a culture temperature of 37°C (p < 0.05). Temperature conditions affected expression levels of Myh (p < 0.0001), Myod1 (p < 0.001), Myog (p < 0.0001), and Mb (p < 0.0001). Oxygen concentration only significantly affected the expression of Myog (p < 0.001). The interaction conditions (Tem × Oxy) had a significant impact on the expression levels of Myod1 (p < 0.001) and Myog (p < 0.0001) (Table 2).
DISCUSSION
Our results showed that heat treatment of C2C12 cells at 39°C inhibited cell proliferation compared to normal temperature and oxygen environment. However, heat treatment at 39°C during differentiation promoted muscle differentiation of C2C12 cells by increasing muscle-related gene expression and protein content. In contrast, oxygen conditions had smaller effects than temperature conditions. In living cells, heat stress is one of the environmental stresses that can negatively affect protein synthesis, protein folding and function changes, and cellular metabolism changes. In addition, it can cause dysfunction of mitochondria of cells and reduce the ability to breathe [29]. Mitochondrial oxidative phosphorylation dysfunction results from the generation of reactive oxygen species (ROS). ROS generation and mitochondrial dysfunction can lead to apoptosis [30,31]. Previous studies have shown that exposure of C2C12 cells to 40°C for 12 to 24 hours can result in accumulation of autophagosomes and increased expression of markers for oxidative stress and apoptosis [32]. In addition, high-temperature heat stress can increase ROS in myoblasts in both cells and induces apoptosis [33]. Therefore, it was considered that heat treatment at 39°C in this study induced apoptosis and the number of cells was low. Since the MTS assay and nuclei number results were similar to cell count results, it could be concluded that heat treatment at 39°C significantly lowered cell number (p < 0.05). When the effect of each condition was confirmed through two-way ANOVA, the temperature of 39°C during C2C12 cell culture negatively affected total number of cells and living cells. Oxygen concentration is considered to have a low effect on cell proliferation. Heat treatment at 39°C and low oxygen concentration showed positive effects on differentiation. Myotube content and fusion index are commonly measured for quantitative evaluation of myotubes [34–36]. The content and maturity of myotubes play a crucial role in determining the efficiency of cultured meat production. [37]. When 39°C and 2% oxygen concentration were applied, the fusion index was significantly higher than that at 37°C and 2% oxygen concentration. The enhancement of myotube area and fusion index appears to have been influenced by an increase in the expression levels of myogenic-related genes or proteins. Myosin is a representative structural protein of muscle cells, serving as a key component of filaments that enable muscle contraction and relaxation [38]. Myogenin is a sequence that causes myoblasts to undergo myoblast differentiation, allowing myoblasts to exit the cell cycle and fuse to form multinucleated myofibrils [39]. MyoD is involved in the specification of myoblasts. It plays an important role in initiating myoblast differentiation [40]. Myoglobin can act as an oxygen transporter in response to mitochondrial demand [41]. Heat treatment at 39°C and hypoxia at 2% induced gene expression level and protein up-regulation of Myoglobin during differentiation. Quinting reported [42] that when Myoglobin was knocked down in cancer cells, the ability to remove ROS and nitric oxide (NO) decreased, leading to an increase in cell necrosis. Similarly to Quinting’s research, this result also suggests that the temperature of 39°C increased ROS, leading to the upregulation of Myoglobin for ROS removal. However, since no experiments related to ROS were conducted in this study, further research is necessary. Based on the above results, it has been shown that a temperature of 39°C during the differentiation process increases the expression levels of Myh, Myog, Myod1, and Mb proteins, thereby promoting differentiation. In this study, the effects of heat treatment at 39°C and hypoxia on the gene expression and protein expression related to the growth and differentiation of muscle cells were confirmed. This suggests that the conditions of 39°C and 2% oxygen may be utilized for efficient cell-cultured meat production.
We investigated effects of temperature and oxygen conditions on cells for technological development of cell cultured meat production. Results of this study showed that heat treatment at 39°C had a negative effect on total cell number and live cell number in cell proliferation compared to normal temperature at 37°C, suppressing proliferation. However, oxygen concentration did not have a significant effect on proliferation. On the other hand, during differentiation, heat treatment at 39°C and hypoxic concentration had a positive effect on myogenic differentiation based on expression levels of Myh, Myod1, Myog, and Mb genes and proteins related to myogenesis and formation. Based on results shown above, it is expected that proliferation proceeds with normal oxygen concentration during cell cultured meat production and that hypoxia and high heat treatment will promote muscle differentiation and have a positive effect in the case of differentiation. However, since this study used a cell line C2C12, additional experiments are needed using primary cells that can be directly obtained from livestock such as Holstein, Hanwoo, and Porcine, which are used for cell-cultured meat. Furthermore, economic aspects related to temperature and oxygen should also be considered when they are directly utilized in cell-cultured meat production.