













Beneficial microorganisms can colonize the host’s intestinal tract and offer benefits to the host due to their unique abilities such as the production of digestive enzymes that enhance feed digestion and absorption [1]. Complex carbohydrate, such as cellulose, cannot be digested by pigs but can only be metabolized by the swine gut microbiota, serving as an important energy source for pigs. Since a significant quantity of cellulose is present in nursery and finisher pig feed, it is desirable to enhance its utilization for improved energy efficiency. As a result, there is a growing interest in researching bacteria associated with cellulose utilization in the swine industry [2], given the substantial cellulose content in swine feed. Insects, such as termites (Isoptera), bookworms (Lepidoptera), and others have been found to harbor symbiotic microflora in their guts, which is responsible for the digestion of cellulosic feed [3]. In this study, we isolated Lactococcus taiwanensis from the gut of a grasshopper (Oxya chinensis sinuosa). It exhibited a low DNA sequence similarity with Lactococcus lactis spp. [4]. Due to its recent discovery, there is relatively limited genomic information available for L. taiwanensis. The aim of this study was to contribute to a more comprehensive understanding of L. taiwanensis at the genomic level.
L. taiwanensis strain K_LL001 was isolated from the gut of a grasshopper (Oxya chinensis sinuosa), collected from the local grasshopper farm in Yangyang, Gangwon-do, Korea. The K_LL001 was grown in MRS broth (BD Difco™, Franklin Lakes, NJ, USA) at 37°C for 24 hours. Genomic DNA was extracted using the MagAttract HMW DNA Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. The complete genome of the L. taiwanensis strain K_LL001 was sequenced using the PacBio RS II (Pacific Biosciences, Menlo Park, CA, USA) platform at Insilicogen (Yongin, Korea). Library preparation was performed using SMRTbell™ Template Prep Kit 1.0 following the manufacturer’s instructions (Pacific Biosciences). A total number of 139,220 long read sequences (854,450,914 base pairs) were produced after subreads filtering sequences. De novo assembly of the gene sequences were performed using the hierarchical genome assembly process (HGAP v2.3.0) workflow, and further polished with Quiver. Quality Assessment Tool for Genome Assemblies (QUAST) v5.2.0 was used for evaluating genome assembly [5]. Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.7 was used for assessing genome completeness and contamination [6]. Prediction of protein coding genes, rRNA and tRNA genes were identified through Rapid Annotation using Subsystem Technology (RAST) server v2.0. Clusters of Orthologous Groups (COGs)-based EggNOG-mapper v2.0 was used to predict functional categorization of protein coding genes [7]. Virulence Factor Database (VFDB) and the Comprehensive Antibiotic Resistance Database (CARD) was used to predict potential virulence factors and antibiotic resistances [8,9]. For the identification of Carbohydrate-Active enzyme (CAZyme), data were submitted to automated carbohydrate-active enzyme and substrate annotation (dbCAN3) website [10].
The complete genome of the L. taiwanensis strain K_LL001 contained one circular chromosome (2,018,259 bp) with a guanine + cytosine (GC) content of 38.75% and no plasmid was found. A total of 2,021 predicted protein-coding sequences, 19 rRNA genes, and 60 tRNA genes were identified in L. taiwanensis strain K_LL001 (Table 1). L. taiwanensis strain K_LL001 had no virulence factor and antibiotic resistance gene. The genome features and genome map of L. taiwanensis strain K_LL001 were illustrated in Fig. 1.
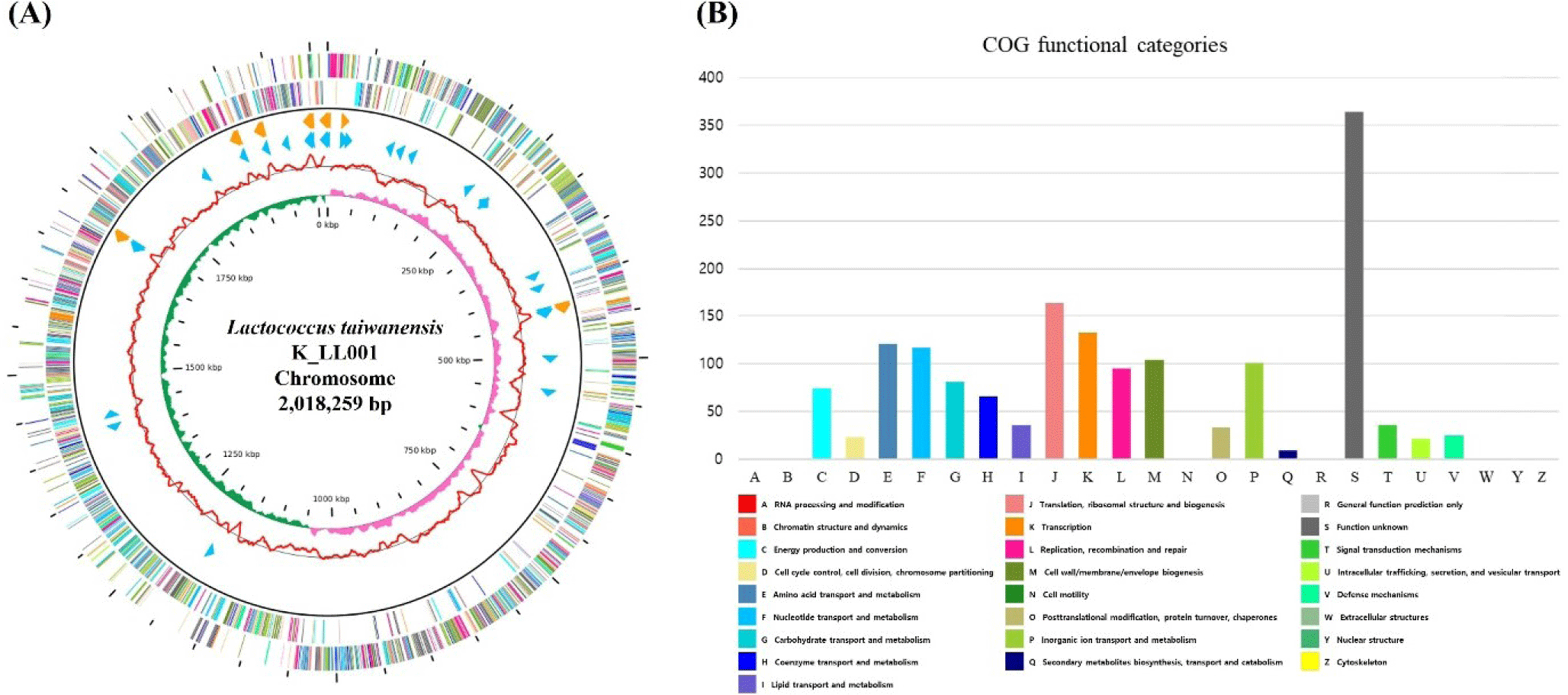
All predicted proteins were subjected to the COG database for functional classification assignment. Top four classifications except S (uncharacterized genes) were J, K, E, and F. The genes categorized under Category J and Category K are associated with translation and transcription of genes in the bacterium. Category E (Amino acid transport and metabolism) and Category F (Nucleotide transport and metabolism) are involved in transport and metabolize amino acids and nucleotides, which are key for bacterial growth and survival.
The summarized distribution of predicted CAZyme was as follows: auxiliary activity (AA) 39; carbohydrate esterases (CE) 34, glycoside hydrolases (GH) 587, glycosyl transferases (GT) 189, carbohydrate-binding module (CBM) 22, and polysaccharide lyases (PL) 2. The CAZyme class mostly possessed in L. taiwanensis strain K_LL001 was GH class. According to Architecture et Fonction des Macromolécules Biologiques (AFMB) laboratory, GHs are the key enzymes involved in carbohydrate metabolism, and they catalyze the hydrolysis of glycosidic bonds in complex carbohydrates such as cellulose, hemicellulose, and starch. In addition, it was verified that L. taiwanensis strain K_LL001 had enzymes which can catalyze the transformation of one glycoside to another. For instance, glycosyltransferase (EC 2.4) is enzyme that catalyze the formation of the glycosidic linkage to form a glycoside, and GH92 family glycosyl hydrolase (EC 3.2.1) genes are a group of glycosyl hydrolases that catalyze the hydrolysis of specific glycosidic bonds in carbohydrates. Moreover, L. taiwanensis strain K_LL001 had no virulence factors or antibiotic-resistant genes. Overall, the genomic characteristics of L. taiwanensis strain K_LL001 suggest that it could be used as probiotics to increase swine performance through enhanced carbohydrate utilization in feed.